Beyond a Single Disease: The Strategic Value of ME/CFS Research
Why ME/CFS Research Informs the Prevention and Understanding of Chronic Disease
Author: Michael Daniels · Framework: GLA Axis — Recovery-Centered Systems Biology (v2.8) · Date: February 10th 2026 · Systems-level research synthesis only. Not medical advice, not a diagnostic tool, and not a claim of clinical validation of any single mechanism.
Scope & framing
This document advances a strategic research claim: ME/CFS is a uniquely high-value
model system in modern biomedicine because it exposes, in accelerated and
observable form, a shared biological failure that underlies many chronic illnesses —
loss of recovery precision.
Rather than being defined by damage to a single organ or ongoing injury, ME/CFS is
characterized by a breakdown in the body’s ability to terminate stress responses
and reliably return to baseline. Individuals may appear relatively stable at rest,
yet exhibit reproducible delayed worsening after physical, cognitive, emotional, or
orthostatic stress, followed by prolonged or incomplete recovery.
Drawing on contemporary aging biology and recent insights into
mitochondria–endoplasmic reticulum contact site (MAM) dynamics,
this work reframes ME/CFS not as a disorder of deficiency or irreversible damage,
but as a failure of dynamic recovery control in which stress responses
initiate appropriately but fail to disengage.
Within this framing, ME/CFS functions as a time-compressed model of chronic
disease persistence and accelerated functional aging. Mechanisms that normally
unfold over decades — declining buffering capacity, impaired termination of recovery
programs, and erosion of resilience — become visible much earlier and more clearly.
What this page does NOT assume:
ongoing viral replication once persistence is established, cytokine storms,
psychogenic causation, or that static baseline biomarkers capture disease severity.
Instead, it argues that the key scientific bottleneck is
recovery execution and termination fidelity across interacting immune,
metabolic, autonomic, vascular, and cellular systems.
Strategic implication:
Because ME/CFS concentrates recovery failure into a measurable phenotype,
it provides disproportionate scientific return. Insights gained here generalize
to Long COVID, infection-associated chronic illness, autoimmune fatigue states,
treatment-related syndromes, and age-associated loss of function — making ME/CFS
research a high-leverage investment for reducing chronic disease burden across
the lifespan.
Abstract
Myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) has historically been treated as a disease defined by symptoms rather than mechanism. Here, we argue that ME/CFS represents a uniquely high-value research model because it exposes, in accelerated and observable form, a shared biological failure that underlies many chronic illnesses: loss of recovery precision.
Drawing on contemporary aging biology and recent insights into mitochondria–endoplasmic reticulum contact site (MAM) dynamics, we reframe ME/CFS not as a disorder of damage or deficiency, but as a failure of dynamic recovery control in which stress responses initiate appropriately but fail to terminate.
We integrate Rattan’s homeodynamics framework, which defines aging as progressive loss of buffering and recovery capacity, with emerging evidence that mistimed and insufficiently reversible MAM-mediated signaling governs recovery execution and healthspan (Rattan, 2014; Monaghan, 2025).
Within this framework, ME/CFS functions as a time-compressed model of chronic disease persistence and accelerated functional aging, offering a powerful lens for understanding how health is lost when recovery fails to disengage.
Studying ME/CFS therefore informs not only treatment of a neglected condition, but the broader prevention and mitigation of chronic disease across the lifespan.
1. Why ME/CFS Research Matters — Far Beyond a Single Disease
Myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) represents one of the highest-value research opportunities in modern biomedicine because it concentrates, in an accelerated and clearly observable form, the same recovery failures that underlie a wide range of chronic illnesses. These include post-viral syndromes such as Long COVID, autoimmune fatigue states, fibromyalgia, post-treatment cancer syndromes, and age-associated frailty. Rather than being defined by damage to a single organ, ME/CFS is characterized by a breakdown in the body’s ability to terminate stress responses and reliably return to baseline—a failure mode increasingly recognized as a shared bottleneck across chronic disease (Unger, 2025; National Academies of Sciences, Engineering, and Medicine, 2024).
Historically underfunded, ME/CFS now occupies a pivotal position at the intersection of infection, immune regulation, metabolism, autonomic control, and aging biology within a single, measurable clinical phenotype. Unlike many chronic conditions that develop slowly and heterogeneously over decades, ME/CFS often follows a discrete triggering event and is marked by reproducible worsening after exertion, delayed symptom amplification, and incomplete recovery. This pattern makes ME/CFS an unusually powerful natural model for understanding how acute stress becomes chronic illness even after the original insult has resolved (Institute of Medicine, 2015).
From a health-science perspective, this aligns closely with contemporary aging biology. Rattan’s homeodynamics framework defines aging not as the accumulation of disease, but as the progressive loss of buffering and recovery capacity. Recent cell biology further identifies mitochondria–endoplasmic reticulum contact sites (MAMs) as central execution surfaces where stress integration, recovery timing, and disengagement are enforced. Within this context, ME/CFS can be understood as a rapid, stress-triggered collapse of recovery execution and timing, rather than irreversible tissue damage—making mechanisms that normally unfold over decades visible much earlier (Rattan, 2014).
Investment in ME/CFS research therefore offers disproportionate return. By targeting failures of dynamic control—how biological systems regulate timing, integration, and termination of recovery—ME/CFS research informs strategies to prevent acute illness from progressing into long-term disability, reduce healthcare burden and workforce loss, and clarify how repeated stress accelerates long-term health decline across systems. Funding ME/CFS research is not a niche or compassionate investment; it is a foundational strategy for reducing chronic disease prevalence, mitigating premature functional aging, and improving resilience across the population.
Supporting context: Broader public-health synthesis and post-acute infection frameworks reinforce this interpretation, including national research-agenda efforts and analyses of unresolved post-infection syndromes (National Academies of Sciences, Engineering, and Medicine, 2024; Choutka et al., 2022).
2. ME/CFS as a High-Value Model of Chronic Disease and Accelerated Aging
ME/CFS offers a rare scientific advantage: it compresses biological processes that typically unfold over decades into a time frame that can be directly observed, measured, and studied. In healthy aging and many chronic conditions, loss of resilience emerges gradually through repeated stress, incomplete recovery, and progressive narrowing of adaptive capacity. In ME/CFS, these same processes appear earlier and more distinctly— often following a single triggering event—making the underlying failure modes unusually clear.
Crucially, ME/CFS does not primarily reflect ongoing injury or cumulative tissue damage. Instead, it reveals what happens when biological systems lose the ability to fully terminate stress responses and return to baseline. Individuals may appear relatively stable at rest, yet experience delayed worsening after physical, cognitive, or emotional exertion, followed by prolonged or incomplete recovery. This pattern closely parallels what aging biology now describes as declining stress tolerance, slower recovery, and reduced buffering capacity—what Rattan conceptualized as progressive loss of homeodynamic space (Wirth & Scheibenbogen, 2020; Scheibenbogen & Wirth, 2024; Charlton et al., 2025).
Recent cell biology further clarifies where this loss of recovery precision is executed. Mitochondria–endoplasmic reticulum contact sites (MAMs) have emerged as central hubs for integrating stress signals and coordinating recovery programs, including calcium signaling, lipid remodeling, redox control, and autophagy. Importantly, chronic pathology is not driven by too little or too much ER–mitochondria contact, but by mistimed persistence and impaired disengagement of these interactions. From this perspective, ME/CFS can be understood as a rapid, stress-triggered collapse of recovery execution and timing at a control surface that normally governs healthspan (Rattan, 2014; Monaghan, 2025).
Because these failures are concentrated, repeatable, and measurable in ME/CFS, the condition functions as a natural stress test of human biology. It allows researchers to study how recovery fails, how resilience erodes, and how chronic control states become self-sustaining—without the confounding effects of decades-long disease evolution. ME/CFS is therefore not an outlier, but an early and accelerated manifestation of mechanisms that ultimately shape many chronic diseases and age-associated decline.
Understanding ME/CFS in this way advances more than disease-specific care. It provides a high-signal model for studying how health is lost through failure of dynamic control—and how restoring recovery precision, rather than compensating for static damage, may be central to preserving function across the lifespan.
Supporting context: Broader systems-aging perspectives reinforce this interpretation, including evidence linking chronic immune activation and inflamm-aging to regulatory imbalance rather than primary damage (Fulop et al., 2018).
Figure — The Recovery Loop Completion
Conceptual model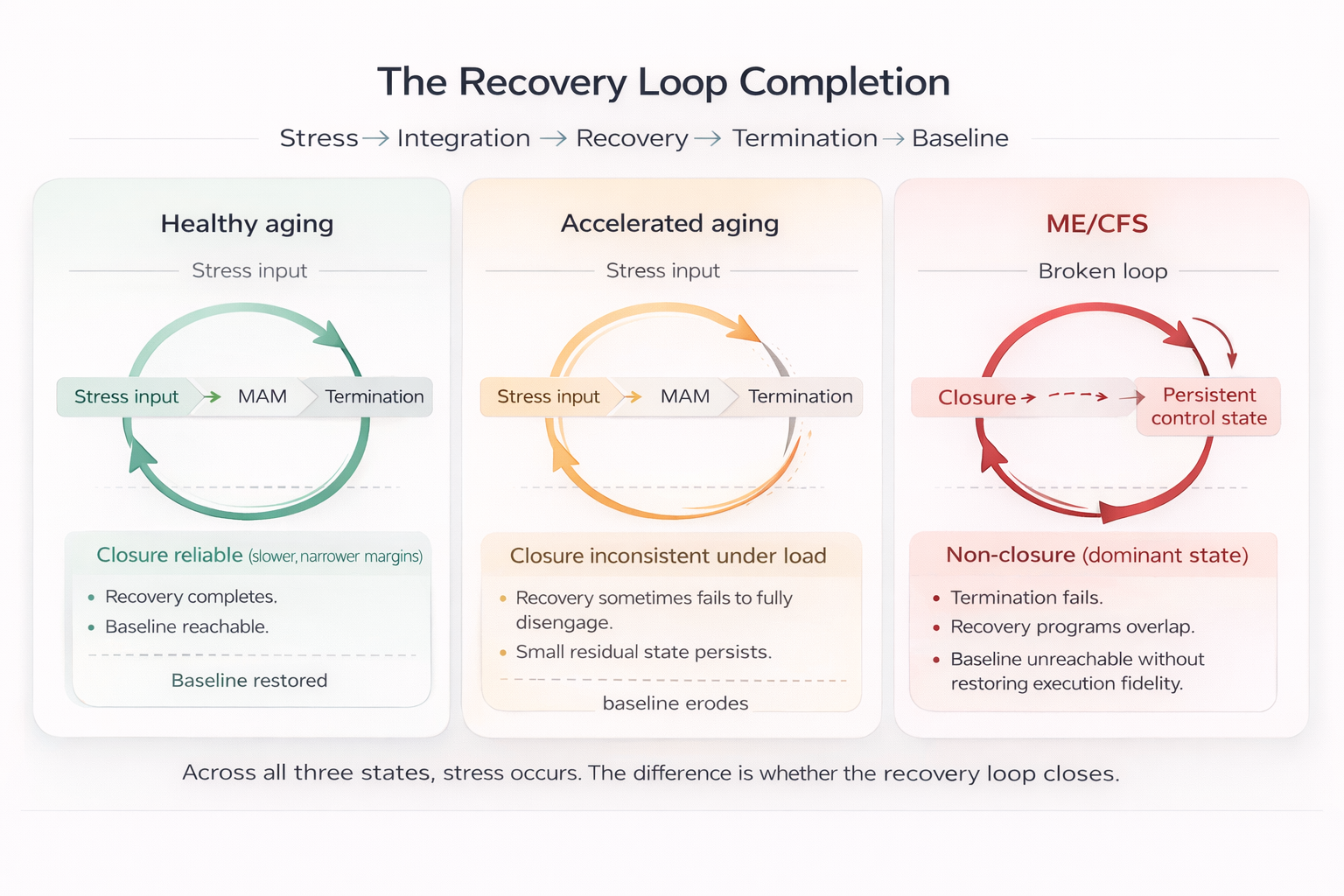
Across all three states, stress occurs. The difference is whether the recovery loop closes. Healthy aging maintains reliable closure; accelerated aging shows inconsistent closure under load; ME/CFS reflects dominant non-closure with persistence. (Rattan, 2014; Monaghan, 2025)
3. Why Traditional Disease Models Fall Short
Most medical research and clinical frameworks are built around identifying ongoing injury, structural damage, or a single malfunctioning system. These approaches work well for acute illness and for diseases in which a clear lesion, pathogen, or failing organ can be identified. However, they struggle to explain conditions in which symptoms persist despite apparent resolution of the original trigger—and in which standard clinical tests often return results within normal ranges.
ME/CFS exposes these limitations with unusual clarity. Individuals may show no evidence of active infection, progressive tissue destruction, or classic inflammatory disease, yet remain profoundly disabled. Attempts to explain the condition through isolated abnormalities—immune activation, metabolic disturbance, autonomic dysfunction, or vascular irregularities—have yielded important observations but have failed to explain why symptoms are delayed after exertion, why recovery is unreliable even with prolonged rest, or why repeated stress progressively lowers tolerance rather than restoring function (Institute of Medicine, 2015; Unger, 2025).
The core limitation of traditional models is that they focus on what is broken, rather than what fails to reset. They are optimized to detect static damage, deficiency, or excess, but not loss of recovery precision or impaired termination of adaptive responses. As a result, they miss failure modes in which biological systems remain structurally intact and appear functional at baseline, yet become unstable under load and unable to return fully to equilibrium.
Contemporary aging biology helps explain this gap. Rattan’s homeodynamics framework emphasizes that health depends on dynamic buffering and recovery capacity rather than static homeostasis, while recent cell biology identifies mitochondria–endoplasmic reticulum contact sites (MAMs) as control surfaces where stress integration, recovery execution, and disengagement must be precisely timed. When these processes become mistimed or insufficiently reversible, adaptive responses persist beyond their useful window, converting reversible stress into chronic dysfunction (Rattan, 2014; Monaghan, 2025).
ME/CFS therefore highlights the need for models that explicitly account for stress-response timing, recovery termination, and the transition from reversible disruption to persistent control states. Without such models, findings remain fragmented, apparent contradictions persist, and interventions fail—not because the biology is unknowable, but because the wrong questions are being asked.
Note: This limitation motivates recovery-centered frameworks that prioritize termination and control dynamics over static damage models, including integrative approaches such as the GLA framework discussed later in this document.
Figure — Timing vs Damage Models (Why Traditional Frameworks Miss ME/CFS)
Model comparisonTwo frameworks can look at the same patient and reach different conclusions. ME/CFS is a high-signal case where dynamic recovery timing matters more than static damage detection.
| Damage-centric model | Timing / termination model (recovery-centered) |
|---|---|
|
Primary question What is broken, injured, deficient, or inflamed right now? |
Primary question What fails to terminate and reliably return to baseline after stress? |
|
Best at detecting Lesions, structural damage, persistent infection, high inflammation, organ failure. |
Best at detecting Delayed symptom amplification, recovery tails, overlap of repair programs, loss of closure fidelity. |
|
Typical evidence Resting/baseline labs, imaging, single time-point biomarkers. |
Typical evidence Load-dependent testing, post-exertional trajectories, repeated measures across time (before/after stress). |
|
Common interpretation “If tests are normal, physiology is normal.” Symptoms without lesions can be misclassified or minimized. |
Common interpretation “Normal baseline can hide instability under load.” Control failure can exist without obvious structural injury. |
|
Typical interventions Treat the detected lesion; suppress symptoms; increase capacity/output. |
Typical interventions Restore termination fidelity; protect recovery bandwidth; reduce overlap of unresolved programs. |
|
Failure mode in ME/CFS Can’t explain delayed worsening (PEM), unreliable recovery, or worsening with “pushing through.” |
Explains Why symptoms can be minimal at rest yet worsen after exertion; why recovery becomes progressively less reliable. |
ME/CFS highlights a gap in damage-centric medical reasoning: many patients show little evidence of ongoing injury at rest, yet exhibit reproducible delayed worsening and incomplete recovery after load. A timing/termination model reframes the core problem as recovery non-closure rather than missing lesions. (Institute of Medicine, 2015; Rattan, 2014; Monaghan, 2025)
4. A Recovery-Centered View of Chronic Illness
A growing body of evidence across medicine indicates that many chronic conditions are not sustained by ongoing injury, but by failure of recovery. In this view, health is not defined solely by the absence of damage, but by the ability of biological systems to return to stability after stress. When recovery is incomplete or imprecise, even normal levels of physical, cognitive, or emotional demand can accumulate lasting effects (Naviaux, 2019).
ME/CFS makes this failure mode visible with unusual clarity. Individuals may tolerate brief periods of activity, only to experience delayed symptom worsening hours or days later, followed by prolonged or incomplete recovery. This pattern reflects a fundamental mismatch between stress response and recovery capacity: the body initiates appropriate responses to demand, but struggles to fully disengage and reset once the demand has passed.
A recovery-centered perspective therefore shifts the focus from identifying isolated abnormalities to understanding how stress responses are terminated, how systems regain balance, and why that process sometimes fails. It explains why symptoms can be absent at rest yet reappear after exertion, why repeated stress progressively lowers tolerance, and why pushing through symptoms often worsens outcomes rather than restoring function.
At the cellular level, this recovery failure reflects impaired execution rather than absent response. Recovery depends on precise execution of programs such as autophagy that are initiated at mitochondria–endoplasmic reticulum contact sites (MAMs), where lipid supply, calcium microdomains, and membrane geometry must be tightly coordinated. Disruption of these contact dynamics does not prevent recovery programs from starting, but impairs their completion, reducing recovery bandwidth and allowing partially resolved cellular stress to persist rather than fully clear (Liu et al., 2025; Janikiewicz et al., 2018).
Importantly, MAM dysfunction in chronic illness is not defined by too little or too much contact, but by mistimed persistence and impaired disengagement of recovery signaling. When MAM-mediated integration fails to terminate appropriately, stress and repair programs overlap and remain partially active beyond their adaptive window, converting reversible stress responses into persistent dysfunction (Monaghan, 2025).
This recovery-failure framing aligns closely with contemporary aging biology. Rattan’s homeodynamics model defines aging as progressive loss of buffering and recovery capacity rather than accumulation of disease, while recent cell biology identifies MAMs as central execution surfaces governing stress integration, recovery timing, and healthspan. Within this framework, ME/CFS can be understood as a rapid, stress-triggered collapse of recovery precision, driven by impaired MAM execution rather than irreversible tissue damage (Rattan, 2014; Monaghan, 2025).
Viewed through this lens, ME/CFS is not an exception to normal biology but an exaggerated, time-compressed example of a general principle: when recovery mechanisms lose precision, health becomes fragile. Studying this failure in ME/CFS provides a powerful opportunity to understand how chronic illness develops, why resilience declines with age and repeated stress, and why protecting recovery—rather than maximizing performance—must be central to preserving long-term health.
Supporting context: The Cell Danger Response framework is complementary here: it helps describe entry into a defensive state, but highlights the importance of understanding why recovery and resolution sometimes fail to complete (Naviaux, 2014; Choutka et al., 2022).
Figure — MAMs as a Recovery Execution Surface
High-level model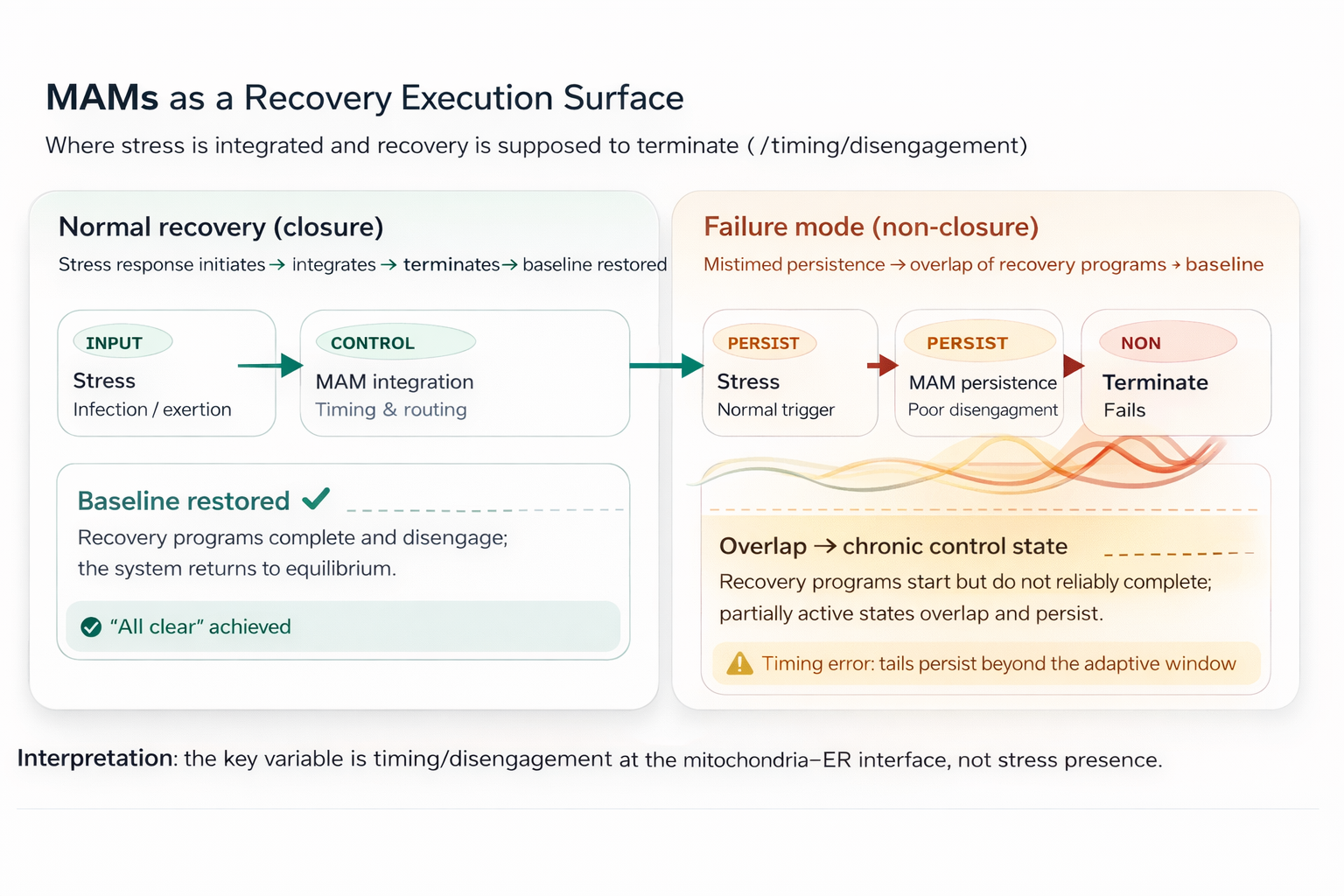
timing schematic: mitochondria–ER contact sites (MAMs) act as an execution surface that integrates stress signals and coordinates recovery programs. Normal closure restores baseline; mistimed persistence produces overlap and a self-sustaining control state. (Monaghan, 2025; Liu et al., 2025)
5. The GLA Framework: Organizing Recovery Failure Across Systems
The GLA framework was developed to address a central unresolved problem in ME/CFS and related chronic conditions: how diverse biological abnormalities converge on the same pattern of failed recovery. Rather than treating immune, metabolic, vascular, autonomic, and neurological findings as competing explanations, GLA organizes them as interacting components of a single recovery system whose stability depends on timing, coordination, and termination (Daniels, 2025–2026).
At its core, the framework focuses on how the body absorbs stress and returns to equilibrium. Many physiological systems can appear intact when assessed in isolation, yet still fail collectively when recovery timing, integration, or buffering capacity is lost. From this perspective, chronic illness does not require ongoing tissue damage or sustained inflammation; it can persist when recovery programs initiate appropriately but fail to disengage, leaving the system vulnerable to repeated destabilization.
A key strength of the GLA framework is its explicit separation of initiation from persistence. Infections, injuries, psychological stressors, or metabolic insults may trigger illness, but they do not by themselves explain why symptoms remain long after those triggers resolve. GLA provides a way to understand how incomplete recovery becomes self-sustaining—why small stressors provoke disproportionate worsening, why symptoms are delayed rather than immediate, and why repeated exertion progressively lowers tolerance instead of restoring function.
Recent cell biology helps localize this failure to a concrete execution surface. Mitochondria–endoplasmic reticulum contact sites (MAMs) have emerged as dynamic communication hubs that integrate calcium, lipid, redox, metabolic, immune, and stress signals to govern cellular recovery and healthspan. In this context, the GLA framework can be understood as a systems-level description of how failures in MAM-mediated signal integration and termination propagate upward into persistent physiological control states (Monaghan, 2025; Liu et al., 2025).
Critically, MAM dysfunction in chronic disease is not defined by too little or too much ER–mitochondria contact, but by mistimed persistence and impaired reversibility of these interactions. When MAM-mediated signaling fails to disengage appropriately, stress and recovery programs overlap and remain partially active beyond their adaptive window, converting otherwise reversible responses into chronic control states. GLA captures how this loss of dynamic control, rather than structural destruction, can explain persistence across ME/CFS, aging-related decline, and other chronic illnesses (Monaghan, 2025).
In this way, the GLA framework does not replace existing disease models. It acts as an integration layer that explains when and why those mechanisms amplify symptoms, why recovery remains unreliable, and how loss of resilience can emerge from timing failure rather than primary damage. By organizing chronic illness around recovery dynamics instead of isolated dysfunctions, GLA offers a coherent architecture for understanding ME/CFS as a disorder of lost control rather than unexplained pathology.
Supporting context: MAM-linked recovery execution is closely tied to maintenance and repair programs (including autophagy initiation and completion) and helps connect cellular execution failure to persistent control states (Janikiewicz et al., 2018).
6. Where the Cell Danger Response Fits — and Where It Stops
The Cell Danger Response (CDR) provides an essential foundation for understanding chronic illness by describing a conserved biological state in which cells prioritize defense and survival in response to perceived threat. During this state, growth, repair, and normal communication are downregulated while protective programs dominate. This framework helps explain why symptoms such as fatigue, hypersensitivity, cognitive slowing, and social withdrawal appear across many illnesses, including ME/CFS (Naviaux, 2014).
CDR is particularly powerful for identifying what state the system has entered. It explains how diverse stressors—such as infection, injury, inflammation, or environmental exposure— can converge on a common defensive response, and why that response is adaptive in the short term. In acute illness, the Cell Danger Response is necessary and protective; effective healing cannot proceed until danger signals diminish and an internal “all clear” is received (Naviaux, 2019).
However, CDR alone does not explain why the system fails to exit this state. It characterizes entry into a defensive mode, but not the mechanisms that govern recovery termination once the original threat has resolved. In ME/CFS, overt danger signals may no longer be present, yet recovery remains incomplete and symptoms persist or worsen with exertion. The critical question is therefore not why defense was initiated, but why it cannot reliably disengage.
The GLA framework extends beyond CDR by focusing on recovery termination and control stability rather than danger activation. It examines how repeated stress, impaired clearance, metabolic strain, and loss of coordination across systems can prevent the body from fully returning to baseline—even in the absence of ongoing threat. In this sense, CDR explains entry into a defensive state; GLA explains how that state can become chronically self-sustaining through failure of recovery execution and timing.
Together, these perspectives are complementary rather than competing. CDR describes the biological logic of defense; GLA describes the systems-level failure that traps the organism in that mode. Understanding both is essential for explaining why ME/CFS persists, why recovery is fragile, and why protecting recovery—rather than pushing performance—is central to preventing long-term decline.
Supporting context: Post-acute infection syndromes and infection-associated chronic conditions highlight the same central gap: explaining persistence after the original insult appears resolved, especially when baseline tests can remain normal (Choutka et al., 2022; Unger, 2025).
7. Why ME/CFS Is a High-Value Research Investment
ME/CFS represents one of the most strategically valuable research opportunities in modern health science because it sits at the intersection of several major public health challenges: post-viral illness, chronic disease persistence, workforce disability, and accelerated health decline. Despite its prevalence and societal impact, ME/CFS has been historically underfunded, leaving fundamental questions about recovery, resilience, and long-term health inadequately addressed (Unger, 2025; National Academies of Sciences, Engineering, and Medicine, 2024; Institute of Medicine, 2015).
Contemporary geroscience increasingly emphasizes that age-related disease risk arises from progressive dysregulation and impaired resolution across physiological systems, rather than linear decline of any single pathway. Because ME/CFS exposes this loss of regulatory balance and recovery precision decades earlier than typical aging, it provides an unusually efficient model for studying the mechanisms that drive both chronic illness and premature functional aging.
Crucially, ME/CFS research targets failures of dynamic control—how biological systems regulate timing, integration, and disengagement of stress responses—rather than static tissue damage or irreversible loss of structure. By focusing on how recovery processes persist, overlap, or fail to terminate, ME/CFS offers a high-leverage entry point for developing interventions that restore regulation and resilience rather than merely compensating for downstream dysfunction.
Research in ME/CFS therefore yields disproportionate return because it addresses shared bottlenecks rather than disease-specific endpoints. Insights gained from studying recovery failure—how systems lose the ability to return to stability after stress—generalize across post-infectious syndromes, autoimmune fatigue states, treatment-related chronic illness, and age-associated loss of function.
ME/CFS also provides a uniquely time-compressed model of chronic disease progression. Processes that typically unfold over decades—declining stress tolerance, impaired coordination across systems, and erosion of recovery precision—are visible much earlier and more clearly. This accelerates discovery by allowing mechanisms of persistence, relapse, and incomplete healing to be studied without the confounding effects of long disease timelines.
From a societal perspective, the stakes are substantial. ME/CFS contributes significantly to long-term disability, healthcare utilization, and loss of workforce participation, particularly among working-age adults. Progress in this field has immediate implications for reducing chronic illness burden, preventing transition from acute illness to long-term disability, and improving population-level resilience to future health stressors.
Funding ME/CFS research is therefore not a niche or compassionate investment. It is an investment in understanding how health is lost—and how it may be preserved—across chronic disease and aging alike. By prioritizing recovery dynamics and control stability rather than isolated symptoms, ME/CFS research offers a high-leverage pathway toward reducing chronic disease prevalence, mitigating premature functional aging, and improving long-term health outcomes at scale.
Mechanistic credibility: Contemporary cell biology supports a recovery-centered interpretation by locating recovery timing, integration, and disengagement at mitochondria–endoplasmic reticulum contact sites (MAMs), linking loss of recovery precision to broader healthspan decline (Monaghan, 2025; Liu et al., 2025; Rattan, 2014). Within the GLA framework (v2.6–v2.8), these findings are organized as a unified recovery-termination failure across systems.
Rattan → GLA → ME/CFS
MAM timing & disengagement framework
| Rattan (2014) Concept | GLA Framing | ME/CFS Interpretation (MAM Timing Updated) |
|---|---|---|
| Homeodynamic space (buffering capacity / resilience) |
Recovery bandwidth | Reduced ability to absorb stress and return to baseline after exertion |
| Shrinkage of homeodynamic space over time | Loss of termination fidelity | Recovery becomes unreliable: delayed worsening, prolonged reset, baseline erosion |
| MARS (maintenance & repair systems) | Recovery execution layers | Recovery programs initiate but do not reliably complete (autophagy / repair / remodeling) |
| Homeodynamics vs homeostasis (dynamic adaptation vs fixed set-point) |
Dynamic control vs static damage | Symptoms reflect failure of control timing rather than irreversible tissue injury |
| Stress response & remodeling are core health functions | Signal-resolution / closure is the bottleneck | Repeated stress overlaps with incomplete recovery, compounding instability |
| MAMs as communication hubs for healthspan (Monaghan, 2025 addition) |
Cellular substrate for recovery-termination failure | Mitochondria–ER contacts integrate recovery signals; dysfunction reflects mistimed persistence and poor disengagement |
| Aging permits disease emergence (aging ≠ disease) | Control failure permits chronic illness | ME/CFS as premature entry into aging-like control states driven by recovery-phase non-closure |
| Health-oriented interventions (maintain resilience) | Restore control before throughput | Therapeutic focus shifts to restoring timing / termination and recovery reliability rather than “pushing energy” |
Rattan provides the homeodynamic buffering framework (Rattan, 2014); Monaghan localizes recovery timing and disengagement to mitochondria–ER contact dynamics (Monaghan, 2025). Mapped using GLA v2.6–v2.8 as an integrative architecture.
Healthy Aging vs Accelerated Aging vs ME/CFS
Contrasted by MAM execution quality
The axis. MAM execution quality refers to how reliably mitochondria–endoplasmic reticulum contact sites coordinate and terminate recovery programs, including:
- calcium microdomain timing and disengagement
- lipid delivery and membrane remodeling at appropriate phases
- redox signaling resolution rather than persistence
- autophagosome initiation and successful completion
Critical clarification: This axis does not describe whether stress responses occur—they do in all three states—but whether recovery programs terminate cleanly and on time, allowing the system to return to baseline.
| State | MAM execution quality (what happens at the mitochondria–ER interface) | Key features |
|---|---|---|
| 1) Healthy aging Gradual narrowing of recovery precision |
In healthy aging, mitochondria–ER contact sites remain functional as recovery execution platforms,
but their precision gradually declines. Autophagy is still initiated and typically completed; calcium
and redox signals are engaged and resolved; and lipid remodeling proceeds in an ordered manner.
However, recovery takes longer, margins narrow, and tolerance for repeated stress progressively decreases.
Importantly, termination still works. Recovery is slower, but not fundamentally unreliable. Unfinished cellular work accumulates slowly, allowing aging to unfold over decades rather than collapsing into persistent illness. |
|
| 2) Accelerated aging Loss of execution reliability under load |
In accelerated aging, MAM execution quality deteriorates more rapidly due to repeated stress, metabolic strain,
inflammation, or genetic susceptibility. ER–mitochondria contacts become maladaptively regulated—not simply reduced
or increased, but mistimed or insufficiently reversible. Autophagy is initiated but more frequently stalls or completes
incompletely, and redox and calcium signals linger beyond their adaptive window.
Recovery still occurs, but with increasing failure rates. Partial resolution becomes common, leading to stepwise baseline erosion rather than smooth decline. Aging-like phenotypes appear earlier than expected, often in midlife rather than late life. |
|
| 3) ME/CFS Recovery failure as a dominant control state |
In ME/CFS, MAM execution quality is degraded to the point that failure of recovery termination becomes the defining
feature, rather than an occasional outcome. Stress responses are initiated appropriately, but autophagy, calcium reset,
redox normalization, and membrane repair frequently fail to complete or disengage. As a result, recovery programs remain
partially active even at rest.
This produces the hallmark clinical pattern: delayed worsening after exertion, prolonged or incomplete recovery, and progressive loss of tolerance. Crucially, this state does not require ongoing tissue damage, persistent infection, or sustained inflammation. It is maintained by repeated recovery attempts that never fully terminate, allowing unresolved cellular work to persist and compound. |
|
The unifying insight
Across all three states, the difference is not the presence of stress, nor the ability to sense or respond to it. The difference is how well recovery is executed and terminated at the mitochondria–ER interface:
- Healthy aging → recovery still closes
- Accelerated aging → recovery closes inconsistently
- ME/CFS → recovery frequently does not close at all
This framing explains why ME/CFS resembles aging biologically yet appears decades earlier—and why the central therapeutic challenge is restoring dynamic control and termination, rather than increasing capacity, suppressing symptoms, or compensating for static damage.
Closing synthesis
Taken together, the evidence presented here reframes ME/CFS not as a disease of damage or deficiency, but as a failure of dynamic recovery control—one that reveals, in compressed form, the same mechanisms that drive chronic disease and functional aging more broadly. By linking Rattan’s concept of homeodynamic capacity with recent insights into mistimed mitochondria–ER contact dynamics, the GLA framework positions ME/CFS as a uniquely informative model for understanding how health is lost when recovery fails to terminate. Studying ME/CFS therefore advances not only treatment for a neglected condition, but the broader science of resilience, recovery, and long-term health preservation.
Conceptual axis adapted from homeodynamics (Rattan, 2014) and updated with mitochondria–ER contact timing and disengagement (Monaghan, 2025). Autophagy and MERC/MAM execution context supported by Janikiewicz et al. (2018) and Liu et al. (2025). Mapped using GLA v2.6–v2.8 as an integrative architecture.
References (APA 7th ed.)
Core framing: Infection-associated chronic illness & public health
Unger, E. R. (2025). Long COVID as an infection-associated chronic condition: Implications. American Journal of Health Promotion, 39(6), 960–965. https://doi.org/10.1177/08901171241308066b PMID: 40485158 · PMCID: PMC12421693
National Academies of Sciences, Engineering, and Medicine; Health and Medicine Division; Board on Global Health; Board on Health Sciences Policy; Committee on Examining the Working Definition for Long COVID. (2024). A Long COVID Definition: A Chronic, Systemic Disease State with Profound Consequences. Washington, DC: National Academies Press. PMID: 39110819
National Academies of Sciences, Engineering, and Medicine; Health and Medicine Division; Board on Health Sciences Policy; Board on Global Health; Forum on Neuroscience and Nervous System Disorders; Forum on Microbial Threats. (2024). Toward a Common Research Agenda in Infection-Associated Chronic Illnesses: Proceedings of a Workshop. Washington, DC: National Academies Press. PMID: 38648305
Committee on the Diagnostic Criteria for Myalgic Encephalomyelitis/Chronic Fatigue Syndrome; Board on the Health of Select Populations; Institute of Medicine. (2015). Beyond Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: Redefining an Illness. Washington, DC: National Academies Press. PMID: 25695122
ME/CFS, post-exertional malaise, and recovery failure
Wirth, K., & Scheibenbogen, C. (2020). A unifying hypothesis of the pathophysiology of myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). Autoimmunity Reviews, 19(6), 102527. https://doi.org/10.1016/j.autrev.2020.102527
Scheibenbogen, C., & Wirth, K. (2024). Skeletal muscle disturbances as a key pathophysiological feature of ME/CFS. Journal of Cachexia, Sarcopenia and Muscle. https://doi.org/10.1002/jcsm.13669
Charlton, B. T., Slaghekke, A., Appelman, B., Eggelbusch, M., Huijts, J. Y., Noort, W., Hendrickse, P. W., Bloemers, F. W., Posthuma, J. J., van Amstel, P., Goulding, R. P., Degens, H., Jaspers, R. T., van Vugt, M., & Wüst, R. C. I. (2025). Skeletal muscle properties in long COVID and ME/CFS differ from those induced by bed rest. medRxiv. https://doi.org/10.1101/2025.05.02.25326885
Cell danger response & failed resolution
Naviaux, R. K. (2014). Metabolic features of the cell danger response. Mitochondrion, 16, 7–17. https://doi.org/10.1016/j.mito.2013.08.006
Naviaux, R. K. (2019). Metabolic features and regulation of the healing cycle—A new model for chronic disease pathogenesis and treatment. Mitochondrion, 46, 278–297. https://doi.org/10.1016/j.mito.2018.08.001
Choutka, J., Jansari, V., Hornig, M., & Iwasaki, A. (2022). Unexplained post-acute infection syndromes. Nature Medicine, 28, 911–923. https://doi.org/10.1038/s41591-022-01810-6
ER–mitochondria (MAM) biology as a chronic-disease control node
Liu, Y., Mao, Z.-H., Huang, J., Wang, H., Zhang, X., Zhou, X., Xu, Y., Pan, S., Liu, D., Liu, Z., & Feng, Q. (2025). Mitochondria-associated endoplasmic reticulum membranes in human health and diseases. MedComm, 6, e70259. https://doi.org/10.1002/mco2.70259
Janikiewicz, J., Szymański, J., Malinska, D., Patalas-Krawczyk, P., Michalska, B., Duszyński, J., Giorgi, C., Bonora, M., Dobrzyn, A., & Wieckowski, M. R. (2018). Mitochondria-associated membranes in aging and senescence: Structure, function, and dynamics. Cell Death & Disease, 9(3), 332. https://doi.org/10.1038/s41419-017-0105-5 PMID: 29491385 · PMCID: PMC5832430
Monaghan, R. M. (2025). The fundamental role of mitochondria–endoplasmic reticulum contacts in ageing and declining healthspan. Open Biology, 15(2), 240287. https://doi.org/10.1098/rsob.240287
Systems framing & accelerated aging
Rattan, S. I. S. (2014). Aging, anti-aging, and hormesis. Mechanisms of Ageing and Development, 140, 1–6. http://dx.doi.org/10.14336/AD
Fulop, T., Larbi, A., Dupuis, G., Le Page, A., Frost, E. H., Cohen, A. A., Witkowski, J. M., & Franceschi, C. (2018). Immunosenescence and inflamm-aging as two sides of the same coin: Friends or foes? Frontiers in Immunology, 8, 1960. https://doi.org/10.3389/fimmu.2017.01960 PMID: 29375577 · PMCID: PMC5767595
Framework reference (this site)
Daniels, M. (2025–2026). GLA Axis: A recovery-centered systems framework for ME/CFS and related chronic illness (v2.6–v2.8). https://mesubtypequestionaire.com
This reference list reflects foundational evidence supporting a recovery-centered, systems-level view of ME/CFS and infection-associated chronic illness. It is not intended as an exhaustive review.
GLA v2.7 — Canonical framework
Current authoritative mechanistic models defining PEM as a recovery-phase failure.
Framework documents
Core architecture and definitions that anchor the GLA model.
Papers
Longer, paper-format documents (reader narrative + figures).
Modules (v2.1 → v2.6)
Modular “building blocks” used across the site. Organized by version and topic.
SMPDL3B phenotype frameworks
Phenotype-specific models (shedding vs deficient) and the mechanistic chain framework.
System modulators & control-state modifiers
Documents that shape interpretation of the core framework and control-state behavior.