Sterol Hinge Timing Module in ME/CFS
A brief overview of the Unified Control State Model (GLA v2.9+): Tier 1 sterol hinge (D > R) shaped by Tier 2 timing modulators and Tier 0 duration inputs
Author: Michael Daniels · Framework: GLA · 2.9+ · Date: February 2026 · Systems-level mechanistic interpretation (not medical advice or treatment guidance).
Scope & framing
The Unified Control State Model (GLA v2.9+) proposes that ME/CFS is a
recovery-phase termination disorder centered on sterol-reset authorization at the
endoplasmic reticulum (ER).
The architecture is explicitly tiered:
• Tier 1 — Sterol Hinge (Acquisition Latch): persistent SCAP–INSIG retention at the ER defines acquisition exclusively when D > R.
• Tier 2 — Timing Layer: ER timing field and nuclear timing gate mechanisms shape sterol exposure timing and effective reset capacity (R) without defining acquisition.
• Tier 0 — Input Suppression Layer: viral persistence, HS-dependent entry, antigen load, and immune complexes alter duration pressure (D) but cannot substitute for the hinge.
Core acquisition criterion (singular):
D > R
Where:
• D = cumulative duration pressure across recovery windows
• R = reset capacity (ability to terminate and reauthorize membrane renewal)
When duration pressure exceeds reset capacity, sterol-reset authorization fails,
SCAP–INSIG retention stabilizes, and membrane renewal remains suppressed.
All immune, metabolic, mechanical, vascular, and viral abnormalities are interpreted
as probability and rate modifiers within this control system. :contentReference[oaicite:2]{index=2}
Core guardrail:
Tier 1 defines acquisition. Tier 2 shapes timing. Tier 0 shapes input load.
No secondary latches are introduced. The hinge remains singular.
Abstract
Myalgic encephalomyelitis / chronic fatigue syndrome (ME/CFS) is characterized by post-exertional malaise (PEM), autonomic instability, and multi-system dysfunction without a unifying mechanistic explanation. The Sterol Hinge Timing Module (GLA v2.9+) presents ME/CFS as a recovery-phase control-state disorder centered on sterol-reset authorization at the endoplasmic reticulum (ER).
The architecture is explicitly tiered. Tier 1 defines the acquisition hinge: sterol-dependent SCAP–INSIG retention at the ER membrane. Disease acquisition occurs exclusively when cumulative duration pressure (D) exceeds reset capacity (R), producing persistent sterol engagement and membrane renewal constraint. No other mechanism substitutes for this definition.
Tier 2 consists of timing-layer modulators that shape how often and how long the hinge is engaged. These include ER sterol exposure timing, endosomal portal bias, oxysterol duty-cycle effects, bile acid phase alignment, nuclear transcriptional persistence, and membrane buffering dynamics. Tier 2 mechanisms alter probability and rate but do not define acquisition.
Tier 0 includes upstream input drivers—viral persistence, heparan sulfate–dependent attachment, antigen load, and circulating immune complexes— that modify duration pressure (D) without independently reversing or defining the hinge.
Within this control-state framework, mitochondrial dysfunction, immune persistence, mechanotransduction instability, lipid remodeling, clearance bottlenecks, and post-acquisition memory states are interpreted as probability and rate modifiers embedded within the system rather than independent disease latches.
ME/CFS is therefore best understood as a sterol-reset authorization failure governed by the inequality D > R. The model reconciles viral persistence, autonomic dysregulation, metabolic instability, and mechanical stress within a single recovery-termination architecture without multiplying disease hinges.
Unified Control State Model of ME/CFS Pathogenesis (GLA v2.9+)
Figure 1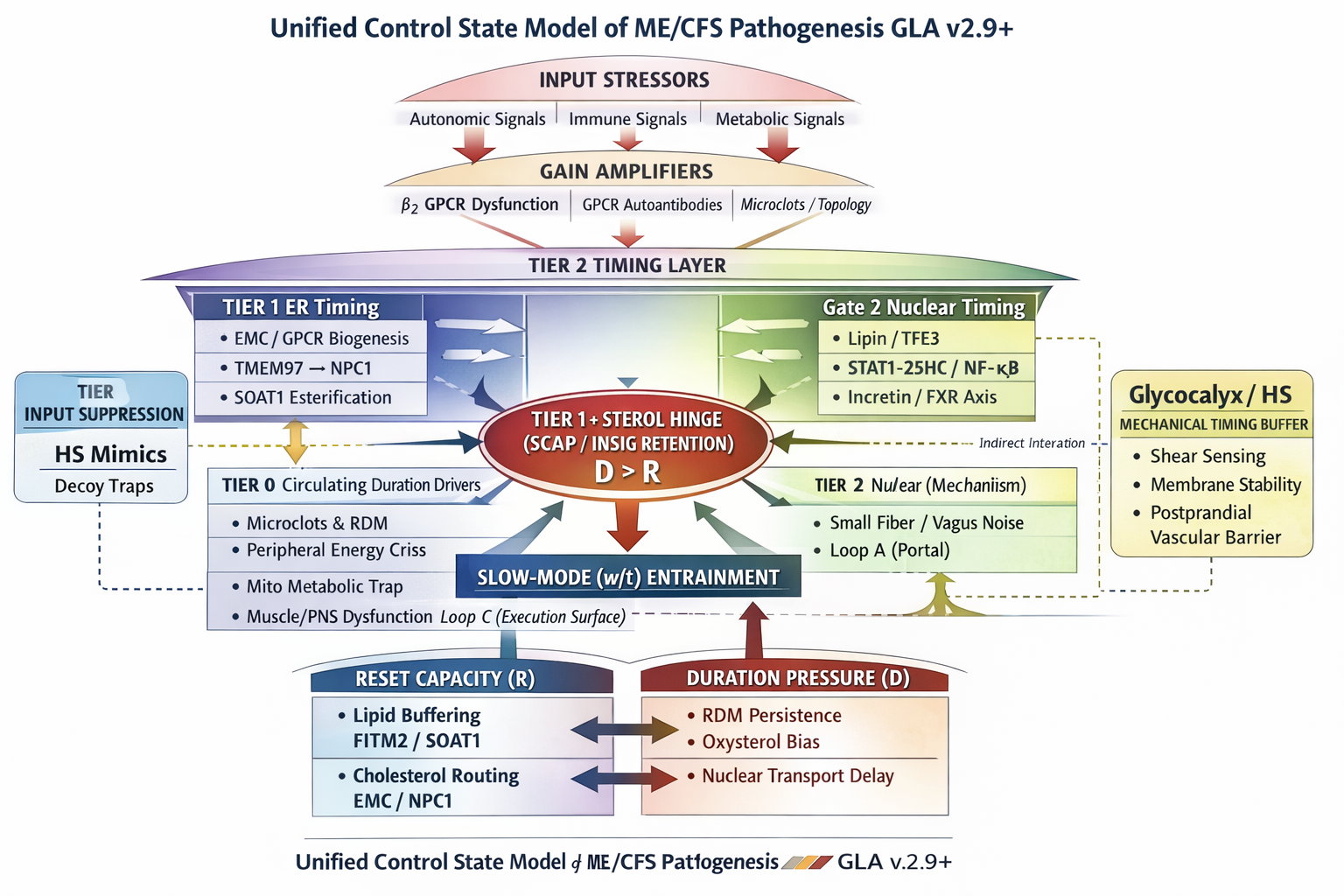
Figure 1. Tiered control architecture of ME/CFS (GLA v2.9+). Tier 1 defines the sterol hinge (SCAP–INSIG retention) crossed only when D > R. Tier 2 timing mechanisms modulate sterol exposure and reset capacity, while Tier 0 inputs modify duration pressure. All immune, metabolic, mechanical, and viral factors operate as probability and rate modifiers within this singular hinge framework.
1. Introduction
Myalgic encephalomyelitis / chronic fatigue syndrome (ME/CFS) is defined clinically by delayed, disproportionate symptom exacerbation following exertion. Traditional models have alternately emphasized immune activation, mitochondrial dysfunction, autonomic dysregulation, viral persistence, or metabolic disturbance. However, none of these models alone explains:
• The non-linear recovery curve
• The coexistence of normal baseline laboratory markers
• The fragility to low physiological loads
The Unified Control State Model (GLA v2.9+) reframes ME/CFS as a control-state disorder characterized by impaired termination of physiological stress responses. Rather than viewing stress magnitude as causal, the model centers on recovery-phase closure fidelity.
The defining problem is not activation itself, but failure of termination. Disease acquisition occurs only when duration pressure exceeds reset capacity.
2. Architectural Overview
The Unified Control State Model is organized into three hierarchical tiers. :contentReference[oaicite:2]{index=2}
Tier 1 — Sterol Hinge (Acquisition Latch)
Tier 1 defines the acquisition hinge. The hinge is governed by sterol-dependent retention of SCAP–INSIG at the ER membrane. When sterol engagement persists across recovery windows such that cumulative duration pressure (D) exceeds reset capacity (R), lipid-reset authorization fails.
D > R
No other mechanism substitutes for this definition. All observed abnormalities must ultimately map to their effect on duration pressure (D) or reset capacity (R).
Tier 2 — Timing Layer (Probability and Rate Modulators)
Tier 2 shapes how often and how long the hinge is engaged. These mechanisms alter exposure timing and reset bandwidth without defining acquisition.
2A. ER Timing Field
• ERC cholesterol redistribution timing
• TMEM97–NPC1 sterol export modulation
• SOAT1 esterification buffering
• Oxysterol bias (e.g., 25-HC)
• Bile acid phase alignment
• Incretin timing (GLP-1 / GIP)
• Glycocalyx mechanical buffering
These mechanisms alter sterol exposure timing without necessarily altering total sterol mass. They modify hinge probability, not hinge identity.
2B. Nuclear Timing Gate
• Lamin A / nuclear periphery sequestration
• Ubiquitin–proteasome turnover
• Chromatin sterol accessibility
These mechanisms regulate transcriptional persistence and effective reset bandwidth (R). They influence how long sterol-reset suppression persists, but do not independently define acquisition.
Tier 0 — Input Suppression Layer
Tier 0 includes upstream drivers that influence duration pressure (D) without defining the hinge:
• Heparan sulfate–dependent viral attachment
• HS mimics (decoy entry inhibitors)
• Antigen load
• Circulating immune complexes
Tier 0 can reduce or increase duration input, but cannot reverse acquisition independently once D > R has stabilized SCAP–INSIG retention.
Tier 2 Timing Modulators vs Tier 1 Sterol Hinge
Figure 2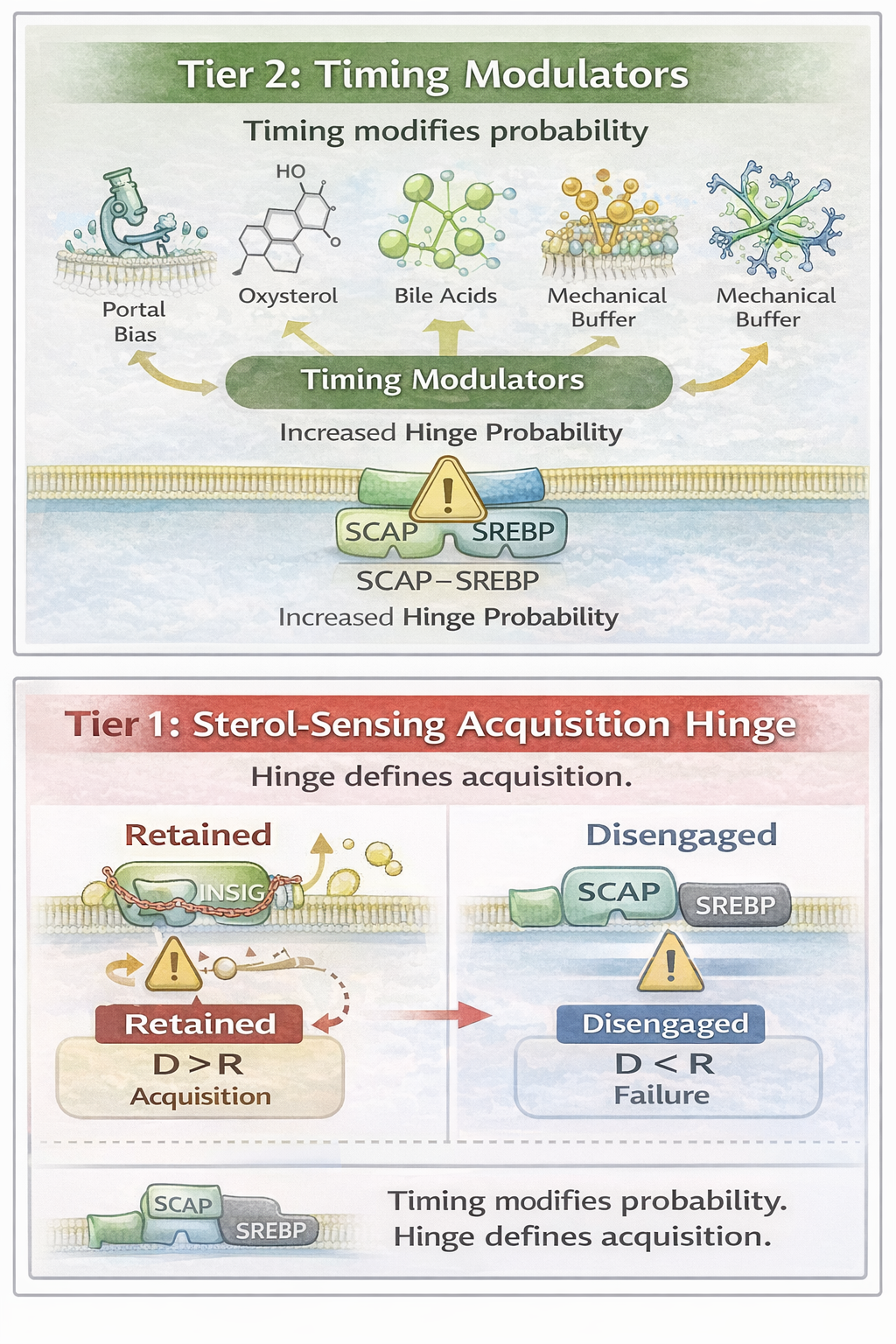
Figure 2. Clear architectural separation between timing-layer modulators and the sterol hinge. Tier 2 mechanisms (portal bias, oxysterols, bile acids, mechanical buffering) modify hinge probability by shaping exposure timing and reset bandwidth. Tier 1 defines acquisition exclusively when D > R. Timing modifies probability. The hinge defines acquisition.
3. Duration Pressure (D) and Reset Capacity (R)
Duration Pressure (D)
Duration pressure (D) represents cumulative persistence of stress-associated signaling across recovery windows. D is kinetic and recovery-phase dependent rather than static. It reflects how long sterol engagement and associated signaling tails remain active.
• Persistent immune signaling (e.g., IFN / STAT)
• Oxysterol-mediated sterol-sensing bias
• Autonomic shear variability
• Mechanical strain on skeletal muscle
• Microclot / topology amplification
• RBC-derived extracellular vesicles
• Glycocalyx degradation
• RDM (Resolution Duration Memory)
These mechanisms do not define acquisition independently. They increase duration persistence and therefore raise the probability that D > R.
Reset Capacity (R)
Reset capacity (R) reflects the system’s ability to terminate sterol engagement and reauthorize membrane renewal between recovery cycles.
• SCAP–INSIG disengagement efficiency
• SREBP processing fidelity
• Lipin nuclear gating
• Proteasomal turnover
• Chromatin permissiveness
• EMC-dependent membrane protein insertion
• FITM2 lipid droplet formation
• SOAT1 esterification buffering
• Sterol trafficking fidelity (NPC1)
• Clearance bandwidth
When reset capacity declines, or duration pressure increases sufficiently, hinge crossing becomes probable.
D > R
Duration pressure increases hinge engagement frequency and persistence. Reset capacity determines whether sterol-reset authorization disengages fully between cycles.
IgG Duration-Driven Sterol Reset Destabilization
Figure 2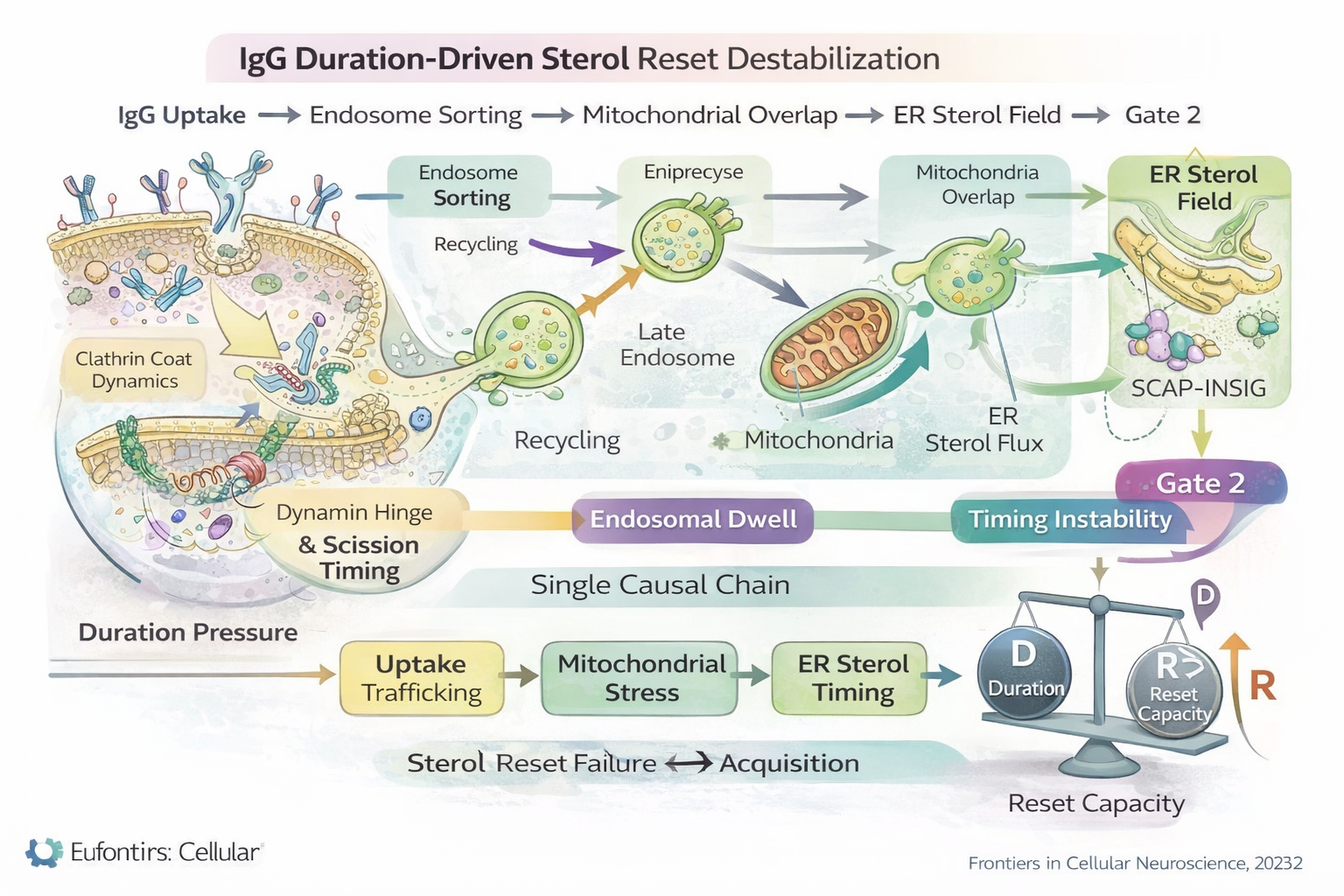
Figure 2. Duration-driven causal chain linking Tier 0 inputs to Tier 1 hinge engagement. IgG uptake increases endosomal dwell time and trafficking variability, producing mitochondrial overlap and ER sterol timing instability. These mechanisms elevate cumulative duration pressure (D) relative to reset capacity (R). Acquisition remains singular and occurs only when D > R at the sterol hinge (Gate 2).
4. Execution Surface: Skeletal Muscle
Skeletal muscle represents the dominant execution surface in which perfusion demand collides with microvascular instability.
During exertion:
• Ca²⁺ flux destabilizes
• ER–mitochondrial redox persistence increases
Importantly, recovery—not exertion—is where collapse occurs. Muscle stress increases duration accumulation by extending recovery tails and increasing slow-mode persistence.
In control-state terms, skeletal muscle stress increases effective duration pressure (D) by prolonging sterol engagement windows. It amplifies hinge probability but does not redefine acquisition.
5. Mechanical Homeostasis vs Fibrotic Positive Feedback
Mechanical stress is not inherently pathological. Within the Unified Control State Model, mechanical modules modify duration pressure (D) and closure probability, but do not independently define acquisition.
Healthy State
• Negative feedback preserves closure probability (p(t))
In a healthy adaptive state, shear sensing and cytoskeletal remodeling restore mechanical equilibrium, limiting recovery tails and preventing cumulative duration persistence.
Pathological State
• Positive feedback stiffening
• Prolonged recovery tails
In the pathological state, mechanical instability becomes self-reinforcing. Shear variability increases persistence windows, prolonging recovery-phase signaling and amplifying duration pressure (D).
Mechanical modules amplify D but do not define acquisition. The hinge remains singular and is crossed only when D > R.
6. GPCR and β₂-Adrenergic Modulation
Functional β₂-adrenergic autoantibodies and GPCR instability may alter signaling gain and closure probability. These mechanisms influence retrigger rate (A(t)) and termination fidelity (p(t)) without redefining the sterol hinge.
• Impaired vasodilatory stabilization
• EMC-dependent GPCR topogenesis affecting receptor stability
Altered β₂ signaling can increase gain and prolong recovery tails, indirectly raising duration pressure (D) by increasing retrigger frequency or reducing closure probability.
These effects remain upstream of the hinge. They shape probability structure but do not substitute for the acquisition criterion D > R.
7. Oxysterols and Immune Licensing
CH25H-derived 25-hydroxycholesterol (25-HC) functions as a sterol-sensing bias ligand, increasing INSIG engagement duty cycle and prolonging sterol retention probability.
• Engagement window widens without necessarily increasing sterol mass
Oxysterol effects are duration amplifiers. They increase the likelihood that sterol-reset authorization remains engaged across recovery windows.
However, oxysterols do not independently define acquisition. The hinge is crossed only when cumulative duration pressure exceeds reset capacity:
Oxysterol bias modifies hinge probability. It does not create a secondary latch.
8. Lipid Droplet and Insertase Axis
EMC-dependent insertion of FITM2 and other membrane clients supports lipid droplet formation and triglyceride buffering. This buffering function helps absorb sterol and redox fluctuations during recovery, reducing ER stress spillover and supporting effective reset capacity (R).
When lipid droplet integrity is impaired, ER stress buffering is reduced and effective R declines. This increases the likelihood that duration pressure (D) will exceed reset capacity across recovery windows.
Lipid droplet / insertase dysfunction modifies R (reset bandwidth). It does not define acquisition independently. The hinge remains singular and is crossed only when D > R.
9. Bile Acid and Incretin Timing
Bile acids and incretin hormones reshape post-prandial sterol flux timing. Within this model, they function as phase modulators that alter when and how sterols are delivered and sensed, thereby influencing hinge probability via timing alignment rather than absolute sterol mass.
By altering sterol exposure timing and transcriptional persistence windows, bile acid and incretin dynamics can increase duration persistence (D) and/or reduce effective reset capacity (R) during vulnerable recovery phases.
Bile acids and incretins influence hinge probability via phase modulation.
They do not directly engage SCAP–INSIG and do not constitute a second hinge.
10. ADIPOSE TISSUE - Resolution Duration Memory (RDM)
After acquisition, Resolution Duration Memory (RDM) sustains an elevated baseline of duration pressure (D) through adipose persistence and clearance bottlenecks. Embedded states can become self-reinforcing, maintaining prolonged recovery tails even when acute inputs are reduced.
RDM is therefore modeled as a post-acquisition persistence substrate: it raises baseline duration load and increases the probability of repeated recovery-phase non-closure, but it does not redefine acquisition.
RDM deepens persistence after acquisition, but the disease state remains defined by the Tier 1 hinge core. Acquisition remains singular and is crossed only when D > R.
11. Therapeutic Implications
Therapeutic strategy must respect the tiered control architecture. Interventions should be classified by whether they lower duration pressure (D), restore reset capacity (R), stabilize gain/termination fidelity, or support buffering capacity.
• D-lowering (e.g., viral entry suppression / input reduction)
• R-restoring (e.g., improving reset bandwidth and reauthorization capacity)
• Gain stabilizers (e.g., autonomic modulation; reducing retrigger likelihood)
• Buffer supports (e.g., lipid droplet integrity; ER stress buffering)
A central sequencing constraint follows directly from the hinge criterion D > R: interventions that increase metabolic or signaling throughput without improving termination fidelity can accelerate duration accumulation and worsen recovery-phase persistence.
Throughput increases without closure restoration risk worsening D.
12. Conclusion
The Unified Control State Model (GLA v2.9+) proposes that ME/CFS is fundamentally a recovery-termination disorder governed by sterol-reset authorization failure. Acquisition occurs exclusively when cumulative duration pressure (D) exceeds reset capacity (R), stabilizing SCAP–INSIG retention across recovery windows. All immune, metabolic, mechanical, vascular, and viral factors are interpreted as modulators of probability and rate within this control architecture, not as independent disease latches.
This model provides a coherent explanation for:
• Non-linear recovery
• Phase-specific fragility
• Heterogeneous therapeutic responses
• Multi-system symptom expression
It unifies mechanotransduction, sterol biology, immune licensing, membrane biogenesis, and clearance dynamics into a single control-state framework without multiplying disease hinges.
Acquisition remains singular and is crossed only when D > R.
Integrated Control-State Pathophysiology (GLA v2.9+)
Figure 3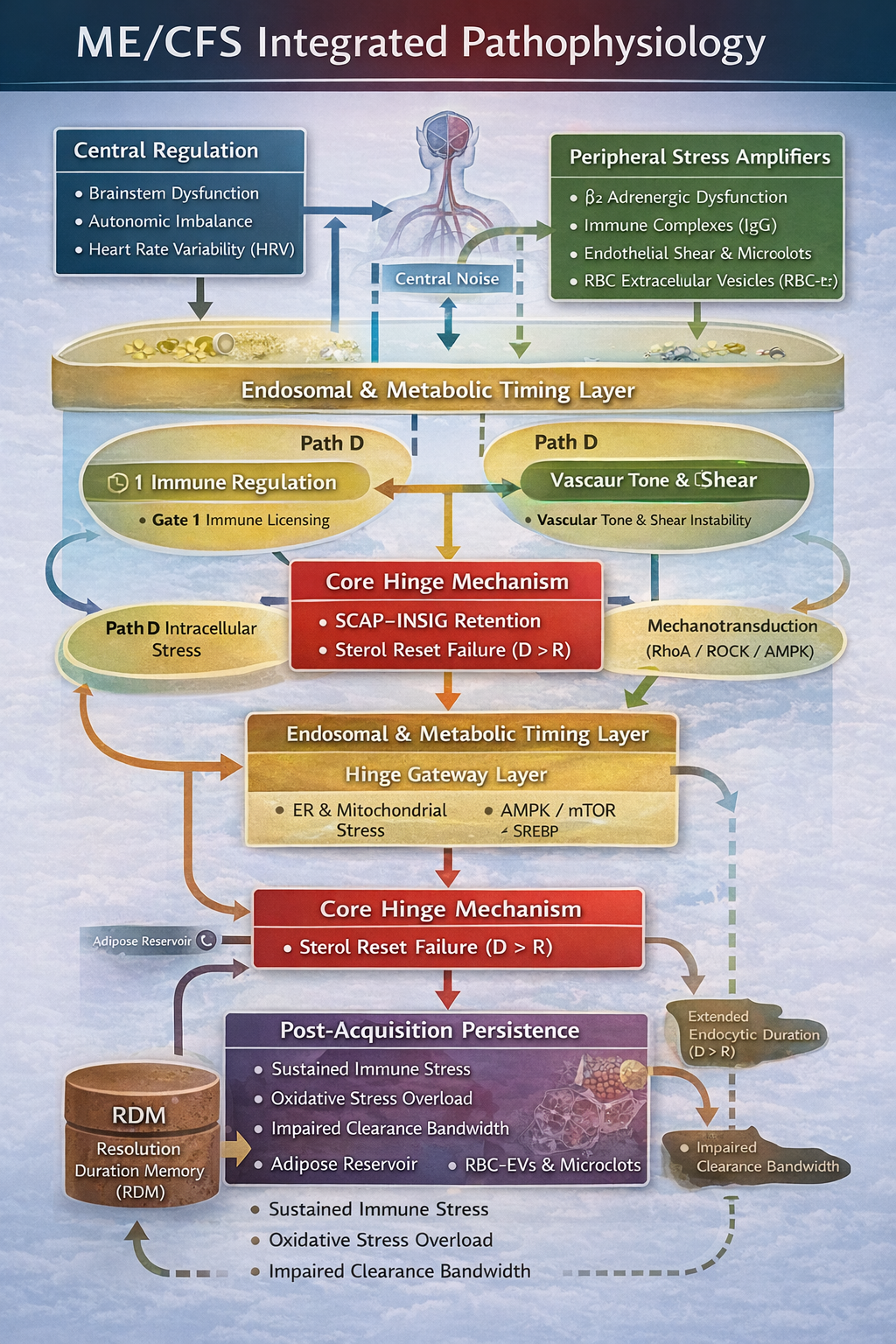
Figure 3. Integrated control-state architecture linking central regulation, peripheral stress amplifiers, endosomal and metabolic timing layers, and the Tier 1 sterol hinge. All upstream immune, vascular, metabolic, and mechanotransductive modules operate as probability and rate modifiers that influence duration pressure (D) and reset capacity (R). Acquisition remains singular and occurs only when D > R, stabilizing SCAP–INSIG retention. Post-acquisition persistence reflects elevated baseline duration load and reduced effective reset bandwidth, not a secondary hinge.
References
Reference list pending
GLA v2.9+ — Canonical framework
Current authoritative mechanistic models defining PEM as a recovery-phase failure.
GLA v2.9+ — Modules
Focused modules expanding Tier 1 hinge logic and Tier 2 timing architecture.
Framework documents
Core architecture and definitions that anchor the GLA model.
Papers
Longer, paper-format documents (reader narrative + figures).
Modules (v2.1 → v2.6)
Modular “building blocks” used across the site. Organized by version and topic.
SMPDL3B phenotype frameworks
Phenotype-specific models (shedding vs deficient) and the mechanistic chain framework.
System modulators & control-state modifiers
Documents that shape interpretation of the core framework and control-state behavior.