The Sterol-Reset Acquisition Hinge in ME/CFS
A brief overview of the single acquisition checkpoint: D > R → sustained SCAP–INSIG retention across recovery windows → suppressed lipid renewal reauthorization
Author: Michael Daniels · Framework: GLA · 2.9 · Date: March 1st 2026 · Systems-level mechanistic interpretation (not medical advice or a treatment recommendation).
Scope & framing
ME/CFS is clinically defined by post-exertional malaise (PEM): delayed, disproportionate symptom worsening following physical, cognitive, orthostatic, or autonomic stress.
In this framework, PEM is not modeled as energy-production deficit or inflammatory excess. Instead, it is modeled as recovery termination failure:
stress responses activate appropriately, but baseline does not reliably re-establish across recovery windows.
This page is a brief overview of the model’s single acquisition checkpoint:
• Loop A — Frequency destabilization: autonomic/mechanical variability increases Ca²⁺ pulse frequency and overlap probability at ER–mitochondrial contact sites.
• Loop B — Duration persistence: biochemical carryover increases time-in-state (Ca²⁺ overlap → redox persistence → immune-linked sterol engagement probability).
• Gate 2 — Sterol-reset authorization hinge: sterol-dependent retention at SCAP–INSIG–SREBP blocks lipid renewal reauthorization when persistence spans recovery windows.
Acquisition condition (singular):
D > R, where D is accumulated duration persistence across recovery windows and R is lipid-reset recovery capacity.
When D exceeds R, sterol engagement fails to disengage between cycles, SCAP–INSIG retention persists, SREBP trafficking remains suppressed, and reversible overlap becomes chemically stabilized non-termination.
What this model does not require:
cytokine storms, persistent viral replication once acquired, systemic hypercholesterolemia as a proxy for ER sterol state, primary global mitochondrial collapse, or psychogenic causation.
The decisive variable is recovery-window kinetics (persistence/overlap), not baseline magnitude.
Core guardrail:
Loop A increases risk but does not define disease. Gate 2 defines acquisition. Upstream factors modify the probability that D > R; downstream layers deepen persistence but do not replace the hinge.
Static baseline measurements alone cannot confirm or refute the hinge — the model requires dynamic recovery-phase sampling.
Sterol-Reset Acquisition Hinge — Overview
Figure 1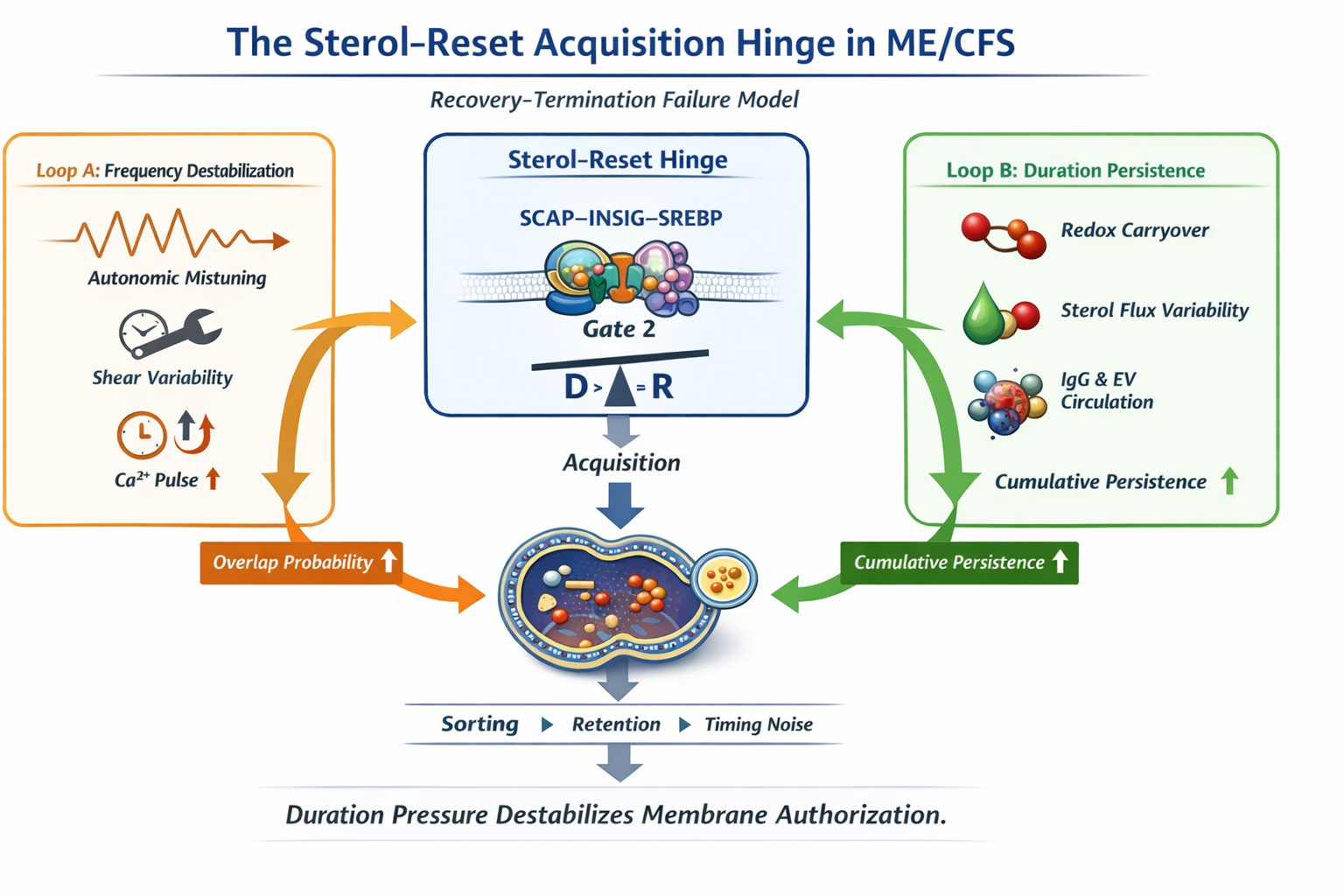
Figure 1. Compact overview of the sterol-reset acquisition hinge. Loop A increases frequency pressure, Loop B increases duration persistence. Acquisition remains singular and occurs only when D > R, producing sustained SCAP–INSIG retention across recovery windows.
1. Overview
Recovery Termination Failure and the Sterol-Reset Acquisition Hinge in ME/CFS
Myalgic Encephalomyelitis / Chronic Fatigue Syndrome (ME/CFS) is clinically defined by post-exertional malaise (PEM), a delayed and disproportionate worsening of symptoms following physical, cognitive, or autonomic stress. In this framework, PEM is not modeled as an energy-production deficit or inflammatory excess. Instead, it is understood as a failure of recovery termination. Stress responses activate appropriately, but the system does not reliably return to baseline.
The model rests on two interacting destabilizing axes:
Loop A — Frequency Destabilization
Mechanical and autonomic variability increase the frequency of intracellular signaling pulses, particularly at ER–mitochondrial contact sites. As pulse frequency rises, the probability of overlap before full decay increases. Loop A increases the likelihood of instability but does not define disease.
Loop B — Duration Persistence
Biochemical carryover prolongs time-in-state. Ca²⁺ overlap, redox persistence, and immune-linked sterol signaling extend the duration of stress-associated engagement. Loop B increases the persistence of signaling, not its magnitude.
Acquisition occurs only when cumulative duration pressure exceeds reset capacity:
D > R
R = lipid-reset recovery capacity
When D exceeds R, sterol-dependent authorization fails to resume. At the molecular level, this corresponds to persistent retention of the SCAP–INSIG complex within the endoplasmic reticulum. Under normal conditions, sterol sensing is transient and membrane renewal reauthorizes between stress episodes. When sterol engagement remains active across recovery windows, membrane lipid renewal is suppressed, and reversible instability becomes chemically stabilized non-termination.
This sterol-reset checkpoint (Gate 2) is singular. It represents the acquisition hinge. Upstream heterogeneity—autonomic variability, immune persistence, lipid remodeling, extracellular vesicle signaling, bile acid timing—modifies the probability that D exceeds R. None independently redefine the hinge.
Downstream of acquisition, additional embedding and rebuild constraints deepen persistence and reduce recovery flexibility. However, these later layers amplify severity; they do not replace the initial authorization failure.
• PEM reflects recovery termination failure.
• Loop A increases frequency pressure.
• Loop B increases duration pressure.
• Acquisition occurs only when D > R and sterol-reset authorization fails.
• Everything upstream modifies probability.
• Everything downstream deepens persistence.
• Gate 2 remains singular.
In this framing, ME/CFS is best understood as a disorder of sterol-dependent recovery authorization—a failure of termination fidelity rather than a failure of energy production.
2. The Acquisition Hinge (Gate 2)
Sterol-Dependent Authorization Failure
The defining transition in this model occurs at a single molecular checkpoint: sterol-dependent authorization failure at the SCAP–INSIG complex within the endoplasmic reticulum (ER). This checkpoint determines whether recovery-phase signaling resolves or becomes chemically stabilized.
Under normal conditions, sterol sensing is dynamic and reversible. SCAP escorts SREBP transcription factors from the ER to the Golgi when sterol levels are low. When sterols accumulate, SCAP binds INSIG proteins, retaining the complex within the ER and suppressing lipid synthesis. This engagement is normally transient. As sterol pressure declines, SCAP disengages, SREBP trafficking resumes, and membrane lipid renewal reauthorizes.
Acquisition occurs when sterol engagement fails to disengage across recovery windows.
2.1 Ligand Amplification: 25-Hydroxycholesterol
25-hydroxycholesterol (25-HC), generated via STAT1-induced expression of CH25H, binds INSIG with high affinity and stabilizes SCAP retention. Physiologically, 25-HC is transient. When duration persistence extends across recovery windows, ligand availability increases the probability that INSIG remains engaged longer than intended.
Ligand elevation alone does not define acquisition. Stabilization occurs only when ligand presence coincides with insufficient reset capacity.
2.2 Reset vs Stabilization
• Temporary INSIG–SCAP retention
• SREBP trafficking pauses
• ER lipid synthesis decreases
• SCAP disengages as sterol pressure resolves
• Membrane renewal reauthorizes
• Baseline is restored
• Sterol engagement persists across recovery windows
• INSIG remains bound
• SCAP–SREBP trafficking remains suppressed
• ERAD pathways are recruited
• Membrane renewal fails to resume
• Recovery termination does not complete
The transition from reversible engagement to stabilized retention defines acquisition.
2.3 The D > R Condition
D > R
D represents cumulative time-in-state of sterol engagement across recovery windows. R represents the system’s ability to restore membrane lipid renewal and disengage INSIG–SCAP retention between stress episodes.
When duration persistence remains below reset capacity, sterol engagement resolves and membrane authorization resumes. When cumulative persistence exceeds reset capacity, disengagement fails to occur before the next stress cycle. Engagement stabilizes. Recovery termination collapses.
• Cytokine storm
• Persistent viral replication
• Global mitochondrial failure
• Systemic hypercholesterolemia
Gate 2 is therefore not gradual metabolic drift but a discrete authorization failure. It is singular and necessary for acquisition.
ME/CFS Integrated Pathophysiology — Hierarchical Architecture
Figure 4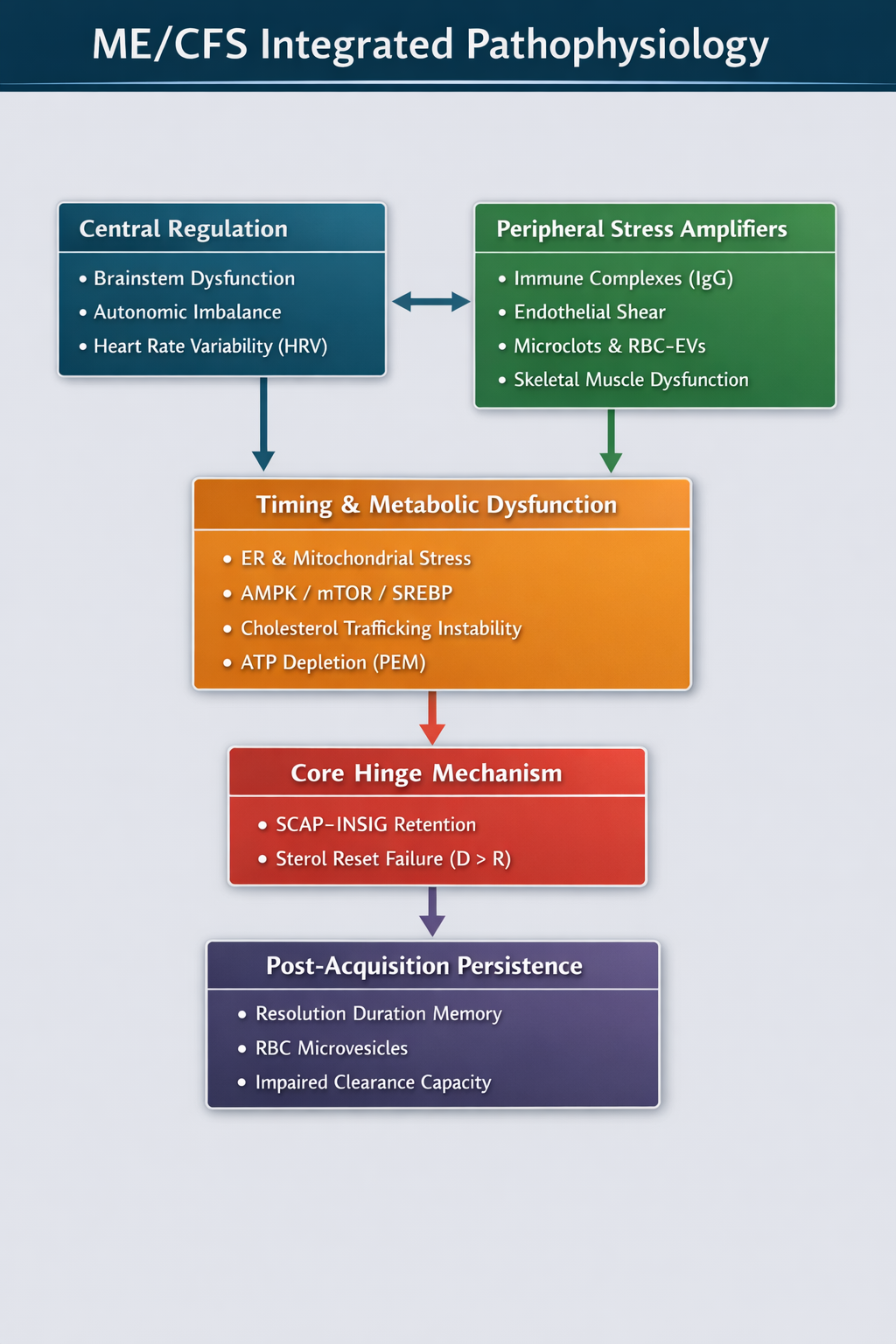
Figure 4. Integrated hierarchy of ME/CFS pathophysiology. Central regulation and peripheral stress amplifiers converge on timing and metabolic instability, including ER–mitochondrial stress and sterol trafficking variability. These upstream pressures increase duration persistence (D) and destabilize reset capacity (R). Acquisition remains singular and occurs only at the sterol-reset hinge when D > R, producing sustained SCAP–INSIG retention. Downstream layers deepen persistence but do not redefine the hinge.
3. Loop A — Frequency Destabilization
Mechanical Overlap Without Chemical Stabilization
Loop A represents the frequency arm of destabilization. It describes how autonomic and mechanical variability increase the probability that intracellular signaling events overlap before full decay. Loop A increases instability probability. It does not define acquisition.
3.1 Autonomic Mistuning and Shear Heterogeneity
Autonomic mistuning introduces increased variability in vascular tone and shear distribution. Microvascular flow becomes spatially and temporally heterogeneous. Shear heterogeneity alters the timing of endothelial mechanotransduction, increasing intracellular Ca²⁺ pulse frequency without requiring amplitude escalation.
3.2 PIEZO1 Timing Noise
Mechanosensitive channels such as PIEZO1 transduce shear into intracellular Ca²⁺ influx. Under heterogeneous shear, activation timing becomes noisy, increasing pulse density rather than sustained concentration.
3.3 Frequency–Decay Relationship
F > Tf
When pulse frequency exceeds decay time, successive pulses overlap before full resolution. Overlap increases partial carryover into subsequent cycles.
At this stage, membrane renewal authorization remains intact. Sterol engagement remains transient. Loop A increases overlap probability but does not stabilize retention.
3.4 Frequency Does Not Define Acquisition
Loop A alone cannot produce acquisition. Even with elevated Ca²⁺ pulse frequency, if duration remains brief and reset capacity intact, recovery termination completes. Loop A increases substrate for Loop B; it does not define the hinge.
4. Loop B — Duration Persistence
Time-in-State as the Destabilizing Variable
Loop B represents the duration arm of destabilization. While Loop A increases frequency, Loop B determines whether events resolve or persist across recovery windows. The destabilizing variable is time-in-state.
4.1 Ca²⁺ Overlap and Redox Persistence
Repeated Ca²⁺ transients increase redox carryover and prolong scaffold activation. Duration increases without requiring amplitude escalation.
4.2 STAT1 Duration and 25-HC Production
Persistent Ca²⁺ overlap increases STAT1 activation, CH25H induction, and 25-HC production. 25-HC binds INSIG and stabilizes SCAP retention. Duration—not magnitude—is the destabilizing variable.
4.3 Duration vs Amplitude
Amplitude-based pathology requires inflammatory escalation. Duration-based pathology requires only that engagement windows remain open long enough to overlap recovery cycles. Acquisition occurs only when cumulative persistence exceeds reset capacity (D > R).
All duration paths converge on sustained SCAP–INSIG retention across recovery windows.
Loop B increases persistence probability.
Gate 2 defines stabilization.
The Five Paths to the Hinge
Parallel Duration Levers
Loop B pressure accumulates through five parallel mechanisms. Each modifies the probability that D exceeds R. None independently define acquisition.
Path A — Ligand Amplification
Persistent STAT1 activity increases CH25H expression and 25-hydroxycholesterol (25-HC) production. Elevated 25-HC increases INSIG binding and stabilizes SCAP retention within the ER.
Path A increases sterol ligand availability. It amplifies engagement probability but remains reversible if reset capacity is sufficient.
Path B — Thiol Disengagement Kinetics
Redox persistence slows thiol-dependent disengagement of signaling scaffolds. Complexes remain in partially activated states longer than intended, widening sterol engagement windows.
Path B increases D without requiring increased ligand supply. It is a termination-precision failure rather than a ligand excess.
Path C — Sterol Partition Bias
Sterol sensing depends on local microenvironment, not total cellular cholesterol. Changes in membrane composition and ER–MAM topology can bias sterol partitioning toward retention-prone configurations.
Path C modifies the spatial sterol field sampled by SCAP–INSIG. It increases the probability that engagement stabilizes under otherwise modest sterol pressure.
Path D — Sterol Flux Timing Variability
Sterol delivery to the ER is temporally regulated. Variability in intracellular cholesterol redistribution introduces timing noise into sterol sensing.
Fluctuations in sterol availability widen engagement windows and reduce reset precision. Path D primarily destabilizes reset capacity (R) by altering sterol-reset timing.
Path E — Circulating Duration Drivers
Systemic inputs, including receptor-bound immunoglobulins and glycome-shifted extracellular vesicles, increase intracellular signaling dwell time and endosomal traffic load.
These inputs do not directly define sterol engagement. Instead, they increase duration pressure upstream, feeding Paths A and B in early phases and sustaining persistence post-acquisition.
Convergence Principle
Sustained SCAP–INSIG retention across recovery windows.
Each path modifies the probability that duration persistence exceeds reset capacity. None independently define acquisition.
Acquisition remains singular and occurs only when:
D > R
Gate 2 defines stabilization.
Figure — Five-Path Convergence
Five-Path Convergence → SCAP–INSIG Retention → D > R
FigureCaption. Loop B duration pressure can rise through five parallel mechanisms (Paths A–E). All converge on a single decision node: sustained SCAP–INSIG retention across recovery windows. Acquisition remains singular and occurs only when D > R.
IgG–Endosome–Mitochondria–Sterol Integration to the Reset Hinge
Figure 2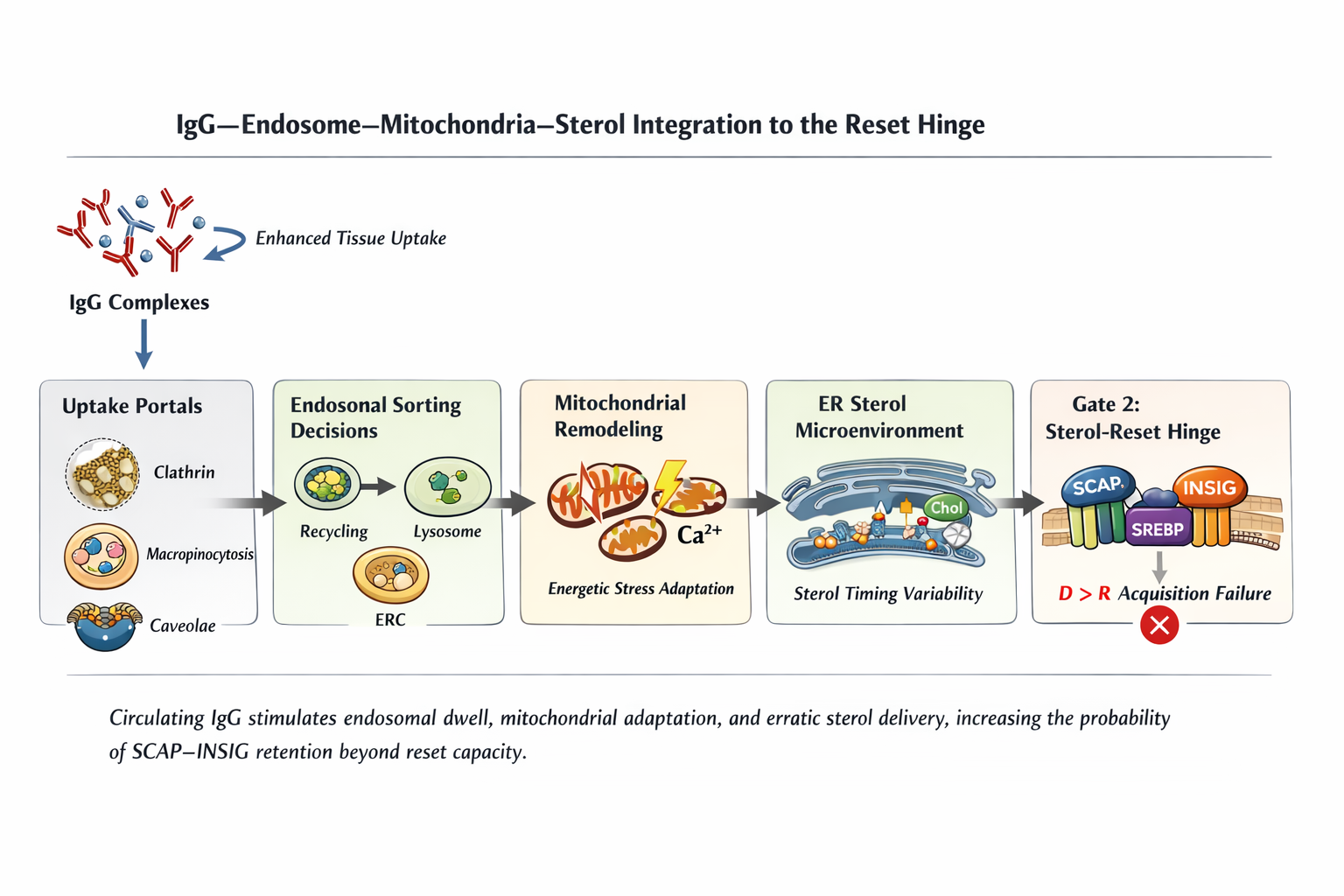
Figure 2. Circulating IgG complexes increase endosomal dwell time and mitochondrial adaptation, introducing sterol flux timing variability within the ER microenvironment. These upstream duration amplifiers increase the probability of sustained SCAP–INSIG retention across recovery windows. Acquisition remains singular and occurs only when D > R.
5. The Endosomal Fate Switch
Routing as a Duration and Reset Modulator
Extracellular duration drivers do not act directly on the sterol-reset hinge. They first enter cells through defined uptake routes and are processed within the endosomal network. The endosomal system functions as a fate-decision layer that determines how long signals persist and how sterol flux is delivered to the ER.
This layer modifies duration pressure (D) and reset capacity (R). It does not define acquisition.
5.1 Three Uptake Routes
• Clathrin-mediated endocytosis (B1) — receptor-coded internalization and signaling tail extension
• Macropinocytosis (B2) — bulk uptake and processing load
• Caveolae-mediated uptake (B3) — mechanosensory and membrane microdomain engagement
Increased receptor occupancy extends endosomal dwell time. Macropinocytosis increases trafficking and degradation load. Caveolae perturbation alters membrane order and shear sensitivity, indirectly influencing signaling frequency.
These routes increase intracellular signaling persistence without necessarily increasing signal amplitude. Entry alone does not determine persistence. The critical transformation occurs during intracellular sorting.
5.2 Sorting Decisions and Sterol Delivery Timing
• Recycling vs degradation
• Early-to-late endosome progression
• Lysosomal fusion and processing
• Recycling compartment redistribution
• Sterol transfer to the ER
These decisions determine the timing and smoothness of sterol delivery to the ER. SCAP–INSIG sterol sensing responds to local sterol microenvironment and temporal fluctuations rather than total cellular cholesterol.
Stable routing preserves phase alignment with recovery cycles. Biased or variable routing introduces sterol flux timing noise, widening sterol engagement windows and increasing the probability that engagement persists across recovery phases.
5.3 Sterol Flux Timing Noise
• Cholesterol export temporally regulated
• Recycling flux balanced
• ER sterol sensing resolves between stress episodes
• Endosomal dwell time increases
• Recycling–degradation balance shifts
• Sterol redistribution pulses become irregular
• ER sterol microenvironment fluctuates unpredictably
Even subtle routing bias is sufficient to alter sterol sensing kinetics. Extreme inflammatory signaling or large cholesterol shifts are not required.
5.4 Hierarchical Clarification
The Endosomal Fate Switch modifies D and R. It increases cumulative duration by prolonging signaling dwell time and can reduce effective reset capacity by destabilizing sterol flux timing. However, acquisition still requires sustained SCAP–INSIG retention when D > R.
Schematic Overview (Condensed)
The Endosomal Fate Switch — Micro-Architecture
Figure 3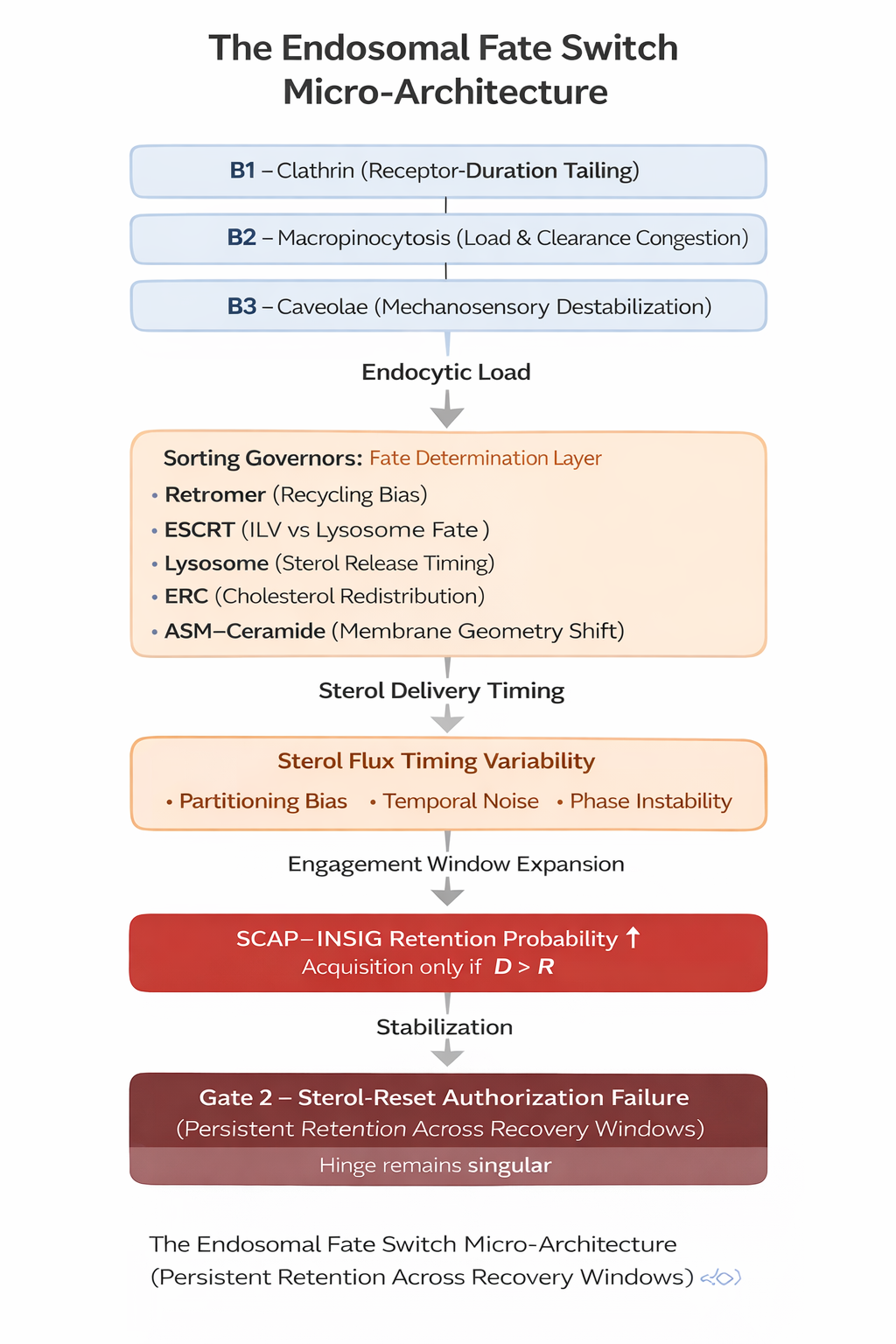
Figure 3. The Endosomal Fate Switch acts as a timing-field modifier upstream of the sterol-reset hinge. Uptake portals (B1–B3) increase endocytic load, sorting governors shape sterol delivery timing, and sterol flux timing variability widens engagement windows. These processes increase the probability of sustained SCAP–INSIG retention, but acquisition remains singular and occurs only when D > R.
6. Bile Acids as Phase Modulators
Sterol Phase Alignment Without Hinge Definition
Bile acids (BAs) influence sterol dynamics at systemic and intracellular levels. Within this framework, they function as timing modulators of the sterol field in which the SCAP–INSIG hinge operates. They do not define acquisition.
6.1 Circadian Sterol Timing
Bile acid synthesis and enterohepatic circulation are intrinsically rhythmic. Oscillatory sterol handling influences the temporal relationship between cholesterol redistribution and cellular recovery cycles.
When well aligned, sterol disposal remains synchronized with stress-resolution phases. When misaligned, sterol flux may occur during vulnerable recovery windows, influencing effective reset capacity (R) without altering total cholesterol mass.
6.2 ERC Cholesterol Extraction
Certain bile acids redistribute cholesterol between compartments, including extraction from the endocytic recycling compartment (ERC) and delivery toward the ER. Temporal variability in this redistribution alters the sterol microenvironment sampled by SCAP–INSIG.
Small ERC↔ER gradient shifts can widen sterol engagement windows even when total cholesterol remains stable.
6.3 Caveolae Modulation
Bile acids influence membrane microdomain organization. Caveolae couple shear sensing to intracellular signaling. Changes in membrane order can increase mechanosensory sensitivity, indirectly raising Loop A frequency and providing substrate for Loop B persistence.
6.4 ASM–Ceramide Remodeling
Bile acids can influence sphingolipid metabolism within late endosomal compartments. Even modest changes in membrane curvature and order can introduce sterol flux variability and increase temporal noise in ER sterol sensing.
Bile acids influence sterol phase alignment, ERC redistribution timing, membrane microdomain stability, and trafficking smoothness.
They modify D and R indirectly.
They do not define the hinge.
7. Phase-Specific Expression
Temporal Dominance Across Acquisition and Persistence
Duration pressures shift across stages of illness, but the acquisition hinge (D > R) remains constant. Upstream inputs modify probability; downstream embedding deepens persistence. The hinge does not change.
7.1 Pre-Acquisition Phase
• Increased signaling duration (D)
• Reset capacity (R) robust
• Sterol engagement transient
• No stable SCAP–INSIG retention
Instability remains probabilistic and potentially reversible.
7.2 Early Acquisition Phase
• Ligand amplification (25-HC)
• Slowed disengagement kinetics
When cumulative duration exceeds reset capacity (D > R), sterol-reset authorization fails. This defines acquisition.
7.3 Embedded State
• Sterol partition bias stabilizes retention-prone microenvironments
• Sterol flux timing variability widens engagement windows
Even modest duration inputs sustain engagement. Embedding architecture maintains instability.
7.4 Advanced State
• Membrane renewal slows
• Redox reserve diminishes
• Lipid-reset cycles lengthen
• Clearance bandwidth decreases
Reduced reset capacity makes persistence self-reinforcing. Gate 5 deepens irreversibility but does not redefine acquisition.
• The sterol-reset hinge remains constant.
• D > R defines acquisition.
• Dominant pressures shift, hierarchy does not.
8. Predictions
Dynamic, Phase-Dependent, and Falsifiable
If ME/CFS represents a sterol-dependent recovery termination failure governed by sustained SCAP–INSIG retention when D > R, then instability should emerge dynamically across a structured stress–recovery sequence (baseline → immediate post-stress → recovery window), rather than as static baseline abnormalities.
The strongest predictions concern sterol-reset timing, bile acid phase alignment, endosomal routing stability, extracellular vesicle identity, and direct INSIG retention dynamics. All predicted abnormalities should manifest primarily during recovery windows, reflecting disproportionate elevation of duration persistence (D) and/or instability of reset capacity (R).
8.1 Dynamic SREBP Suppression During PEM
During post-exertional malaise (PEM), sterol-reset instability should be detectable as prolonged suppression of SREBP processing relative to controls.
• Reduced SREBP2 cleavage
• Sustained suppression of sterol-responsive transcription
• Delayed normalization of lipid-synthesis gene expression
These changes should:
• Correlate with symptom severity
• Resolve more slowly in patients than in controls
Closure probability (p(t)) should remain suppressed, and termination hazard (h(t)) reduced during recovery windows. If sterol engagement does not persist abnormally during PEM, the sterol-reset hinge hypothesis fails.
8.2 Bile Acid Timing Correlates With Symptom Fluctuation
Symptom variability should correlate more strongly with bile acid timing than with total bile acid concentration.
• Greater symptom amplification following large or late meals
• Altered post-prandial bile acid pulse timing
• Increased sterol flux variability during post-prandial recovery windows
Total bile acid levels may remain within normal range. The relevant variable is phase alignment relative to recovery cycles. Bile acid timing should measurably alter effective reset capacity (R) or widen sterol engagement windows, thereby increasing the probability that D > R.
If bile acid timing does not measurably alter recovery-phase sterol-reset dynamics, their role as phase modulators must be reconsidered.
8.3 Endosomal Routing Variability During Recovery
Endosomal stress and routing markers should change specifically during recovery windows, not merely at baseline.
• Altered recycling–degradation balance
• Increased variability in endosomal maturation markers
• Subtle lysosomal acidification instability
• Increased cargo dwell-time during recovery
These routing shifts should correlate temporally with post-exertional fatigue and cognitive worsening.
Routing variability is predicted to increase duration persistence (D) and/or destabilize reset capacity (R) by introducing sterol flux timing noise. However, routing instability does not redefine the hinge.
If sustained SCAP–INSIG retention occurs in the absence of routing instability, routing must be considered a probability modifier rather than a necessary contributor.
8.4 EV Glycome Shifts During Stress Windows
Extracellular vesicle (EV) identity should shift dynamically during stress and recovery phases.
• Increased mannose-enriched EV fractions during recovery
• EV cargo enriched for JAK–STAT and ER stress signatures
• Correlation between EV glycome shifts and symptom severity
These shifts should reflect duration amplification and dwell-time extension rather than cytokine amplitude escalation. EV-related inputs should increase D by prolonging intracellular signaling persistence, without independently defining acquisition.
If EV identity remains static across stress cycles, Path E must be reweighted as a probability modifier.
8.5 INSIG Retention Persistence
The most central and decisive prediction concerns SCAP–INSIG retention.
• INSIG occupancy should remain elevated longer in ME/CFS than in controls
• Sterol-responsive gene suppression should persist beyond expected resolution windows
• Reset timing should be measurably delayed
Direct measurement of SCAP trafficking dynamics or INSIG retention duration across recovery windows would provide decisive validation.
If INSIG retention does not persist abnormally during recovery, the sterol-reset acquisition model collapses.
Integrated Prediction Logic
If the model is correct, then during recovery:
• Effective reset capacity (R) declines or becomes unstable
• Closure probability (p(t)) decreases
• Termination hazard (h(t)) remains suppressed
• Sterol microenvironment variability widens engagement windows
• Routing instability correlates with symptoms
• Bile acid phase alignment modulates symptom intensity
These abnormalities should be dynamic and phase-dependent, not static baseline elevations.
Acquisition remains defined solely by sustained SCAP–INSIG retention when D > R.
9. Therapeutic Sequencing
Control Precedes Capacity
If ME/CFS represents a sterol-reset acquisition disorder governed by the inequality D > R, then therapeutic strategy must follow the architecture of instability. Interventions that increase metabolic throughput in an unstable control system risk increasing duration persistence (D), lowering closure probability (p(t)), and reinforcing sustained SCAP–INSIG retention.
Control precedes capacity.
Termination fidelity must improve before energetic throughput increases.
Therapeutic sequencing must therefore respect the hierarchical structure of the model. No intervention substitutes for restoration of sterol-reset authorization timing.
9.1 Stabilize Loop A First (Frequency Control)
Loop A increases signaling overlap probability by elevating retrigger rate (A(t)). Early intervention should reduce frequency instability before attempting to increase energetic output.
• Reduce shear variability and autonomic mistuning
• Lower Ca²⁺ pulse frequency density
• Restore spacing between signaling events
When frequency stabilizes:
• Duration accumulation (D) slows
• Reset capacity (R) is less likely to be exceeded
Increasing metabolic drive while Loop A remains unstable increases retrigger rate (A(t)), accelerating duration accumulation and raising the probability that D > R.
Preventive note:
Heparan sulfate (HS) mimics and glycocalyx-stabilizing strategies act at this level.
By improving endothelial shear filtering and mechanotransduction precision,
they reduce frequency-driven overlap and therefore lower the probability of
crossing the acquisition threshold.
HS mimics are therefore best understood as preventive stabilizers,
not hinge reversers.
9.2 Reduce Duration Pressure (Loop B)
After frequency stabilization, attention shifts to reducing duration persistence and improving disengagement kinetics.
• Shorten signaling dwell time (reduce D)
• Improve thiol-dependent disengagement precision
• Normalize immune-duration tails
• Narrow sterol engagement windows
The goal is to raise effective closure probability (p(t)) and increase termination hazard (h(t)) during recovery windows.
Loop B reduction decreases the probability that sterol engagement persists long enough to prevent membrane renewal reauthorization.
9.3 Protect Sterol Authorization Timing (Gate 2 Preservation)
Once frequency and duration pressures are reduced, interventions may focus on protecting sterol-reset timing precision.
• Preserve SCAP–INSIG disengagement kinetics
• Protect membrane lipid renewal authorization
• Stabilize ER sterol microenvironment timing
The objective is not to override sterol sensing, but to ensure that engagement remains transient and resolves fully between recovery windows.
If sterol-reset authorization is not restored, increasing mitochondrial throughput risks deepening retention by increasing metabolic signaling density.
9.4 Correct Embedding (Paths C and D)
After authorization timing improves, embedding layers that reinforce persistence can be addressed.
• Improve membrane structural stability
• Reduce sterol flux timing noise
• Improve endosomal routing smoothness
• Reduce retention-prone microenvironment bias
These interventions modify probability structure by stabilizing effective reset capacity (R). They do not redefine the hinge.
9.5 Rebuild Reset Capacity (Gate 5)
In advanced states, reset capacity (R) becomes intrinsically reduced.
• Restore membrane renewal bandwidth
• Improve redox reserve
• Increase recovery robustness
• Lengthen recovery-window closure probability
Only once effective R is improving should significant metabolic throughput be increased.
9.6 Sequencing Summary
1. Stabilize frequency (Loop A)
2. Reduce duration pressure (Loop B)
3. Preserve sterol authorization timing (Gate 2 protection)
4. Correct membrane and routing embedding (Paths C + D)
5. Rebuild reset capacity (Gate 5)
6. Only then increase metabolic throughput
Throughput introduced before termination fidelity is restored risks increasing D faster than R can recover.
This sequencing preserves the singular hinge. It does not treat symptoms in isolation. It restores termination precision first.
ER Stress / UPR → MAM Timing → Sterol Reset (Gate 2) — Outcome States
Figure 5Figure 5. Outcome-state contrast across stress and recovery. Healthy adaptation resolves ER stress/UPR, restores MAM timing, and fully reauthorizes sterol reset (Gate 2). Under reversible overload, ER stress and sterol engagement can increase transiently, but resolution occurs if recovery completes. In ME/CFS persistence, recovery termination fails: duration persistence (D) remains elevated and reset capacity (R) becomes insufficient, increasing the probability of sustained SCAP–INSIG retention across recovery windows. Acquisition remains singular and occurs only when D > R; downstream persistence reflects reinforcement of the acquired state rather than a second hinge.
Gut Viral Persistence as a Duration Driver → From Input to Sterol-Reset Failure (D > R)
Figure 6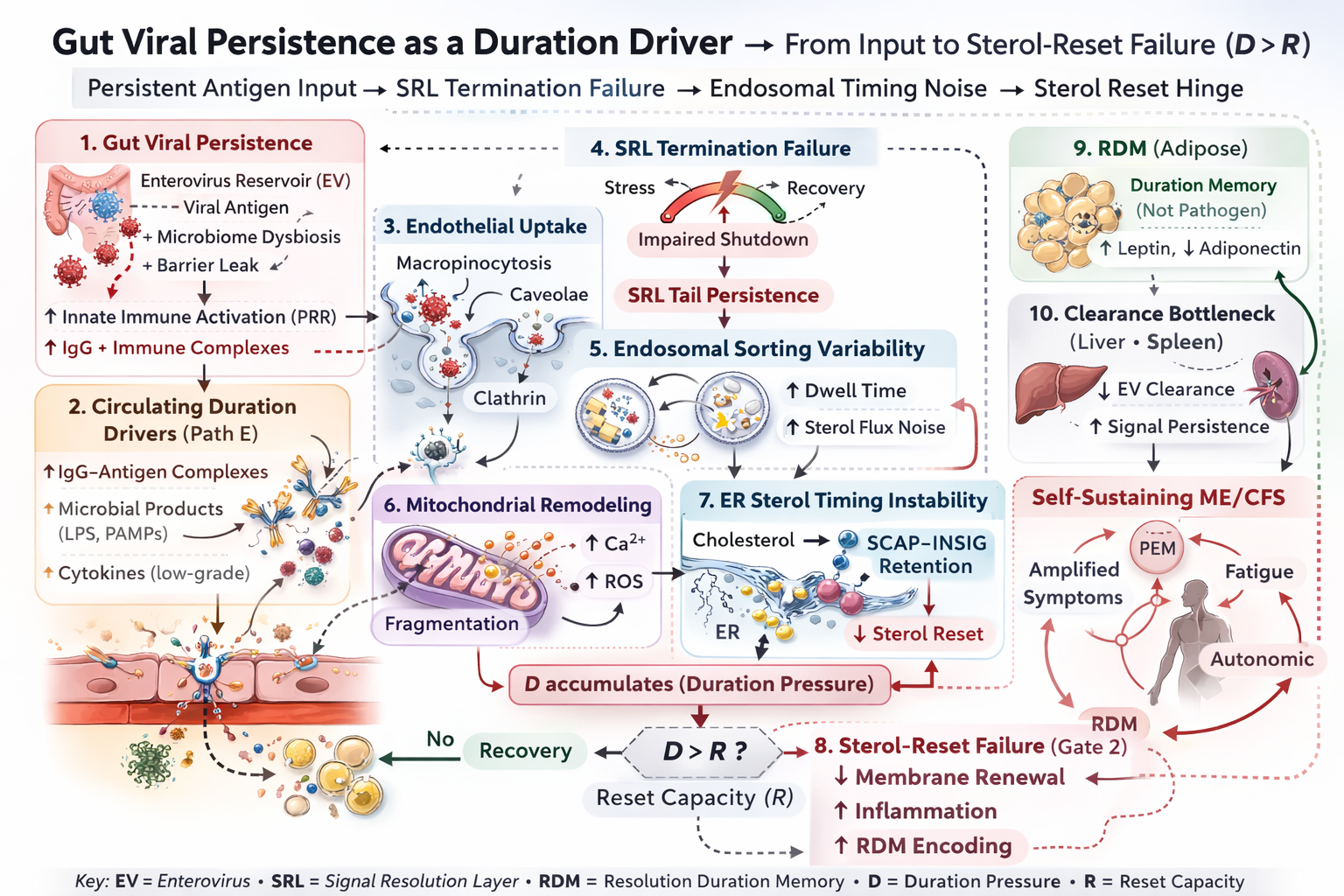
Figure 6. Candidate upstream duration-driver pathway: persistent antigen input biases signal resolution failure (SRL tail persistence), increases endosomal timing noise and sterol flux variability, and elevates cumulative duration pressure (D) while destabilizing effective reset capacity (R). These effects increase the probability of sustained SCAP–INSIG retention across recovery windows. Acquisition remains singular and occurs only when D > R at Gate 2; downstream persistence layers amplify and stabilize the acquired state but do not redefine the hinge.
10. Concluding Synthesis
Sterol-Reset Failure as a Unified Recovery-Termination Mechanism (v2.9+)
Myalgic Encephalomyelitis / Chronic Fatigue Syndrome (ME/CFS) is clinically defined by post-exertional malaise (PEM), a delayed worsening of symptoms following physical, cognitive, or autonomic stress. Within this framework, PEM is not interpreted as primary energy-production failure, inflammatory excess, or structural degeneration. It is defined as a failure of recovery termination. Stress responses activate; closure probability declines; termination does not complete.
Instability emerges from two interacting pressures. Loop A increases retrigger rate (A(t)) through autonomic mistuning and mechanical overlap. Loop B increases duration persistence (D) through Ca²⁺ carryover, redox persistence, ligand amplification, sterol partition bias, sterol flux timing variability, and circulating duration drivers.
These pressures converge at a single molecular checkpoint: sterol-dependent authorization at the SCAP–INSIG complex. This is the sole acquisition hinge (Gate 2).
Acquisition occurs only when D > R.
D represents cumulative duration persistence across recovery windows. R represents effective reset capacity — the ability to disengage sterol sensing and reauthorize membrane lipid renewal between stress cycles.
When sterol engagement fails to disengage across recovery windows, SCAP–INSIG retention persists, SREBP trafficking remains suppressed, closure probability (p(t)) remains reduced, termination hazard (h(t)) remains suppressed, and membrane renewal does not reauthorize.
Reversible signaling overlap becomes chemically stabilized non-termination. The hinge remains singular. Everything upstream modifies probability structure. Everything downstream deepens persistence depth and rigidity.
Tier 1 (Hinge): Binary sterol-reset authorization state (retained vs reauthorized).
Tier 2 (Timing Field): Portal bias, routing variability, bile acid phase alignment, sterol microenvironment fluctuations — these refine D, R, and p(t), but do not constitute a second hinge.
This architecture explains why:
• Biomarkers appear modest at rest but destabilize dynamically across recovery windows
• Post-prandial states influence symptom severity via sterol phase alignment
• Endosomal routing, lipid remodeling, extracellular vesicle identity, and bile acid timing converge without requiring cytokine storm or global mitochondrial collapse
• The phenotype reflects timing instability rather than intensity excess
Crucially, the model is hierarchical and falsifiable. If sustained SCAP–INSIG retention does not occur abnormally during recovery windows, or if closure probability and sterol-reset timing normalize appropriately, the hinge hypothesis collapses.
Conversely, direct demonstration of prolonged INSIG occupancy and delayed sterol-reset disengagement during recovery would provide decisive validation.
ME/CFS is therefore best understood as a sterol-reset acquisition disorder in which cumulative duration pressure destabilizes membrane authorization across stress–recovery cycles.
The disease reflects a failure of membrane-based termination fidelity — a breakdown of sterol-phase alignment that transforms transient stress overlap into persistent non-closure.
References
APA 7th Edition
- Hanson, M. R. (2023). The viral origin of myalgic encephalomyelitis/chronic fatigue syndrome. PLoS Pathogens, 19(8), e1011523. https://doi.org/10.1371/journal.ppat.1011523
- Glass, K. A., Giloteaux, L., Zhang, S., & Hanson, M. R. (2025). Extracellular vesicle proteomics uncovers energy metabolism, complement system, and endoplasmic reticulum stress response dysregulation postexercise in males with myalgic encephalomyelitis/chronic fatigue syndrome. Clinical and Translational Medicine, 15(5), e70346. https://doi.org/10.1002/ctm2.70346
- Liu, Z., Hollmann, C., Kalanidhi, S., Lamer, S., Schlosser, A., Basens, E. E., Nikolayshvili, G., Sokolovska, L., Riemekasten, G., Rust, R., Bellmann-Strobl, J., Paul, F., Naviaux, R. K., Nora-Krukle, Z., Sotzny, F., Scheibenbogen, C., & Prusty, B. K. (2026). Immunoglobulin G complexes from post-infectious ME/CFS, including post-COVID ME/CFS, disrupt cellular energetics and alter inflammatory marker secretion. Brain, Behavior, & Immunity – Health, 52, 101187. https://doi.org/10.1016/j.bbih.2026.101187
- Missailidis, D., Armstrong, C. W., Anderson, D., Allan, C. Y., Sanislav, O., Smith, P. K., Esmaili, T., Creek, D. J., Annesley, S. J., & Fisher, P. R. (2026). Multi-omics identifies lipid accumulation in myalgic encephalomyelitis/chronic fatigue syndrome cell lines: A case-control study. Journal of Translational Medicine, 24(1), 145. https://doi.org/10.1186/s12967-025-07620-x
- Pesqueira Sanchez, M. A., de Necochea Campion, R., Dalhuisen, T., Fehrman, E. A., Chhabra, P. S., Kelly, J. D., Martin, J. N., Deeks, S. G., Henrich, T. J., Peluso, M. J., & LaRosa, S. P. (2025). Increased mannosylation of extracellular vesicles in long COVID plasma provides a potential therapeutic target for Galanthus nivalis agglutinin (GNA) affinity resin. bioRxiv. https://doi.org/10.1101/2025.11.21.689519
- Alfred, V., & Vaccari, T. (2016). When membranes need an ESCRT: Endosomal sorting and membrane remodelling in health and disease. Swiss Medical Weekly, 146, w14347. https://doi.org/10.4414/smw.2016.14347
- Bramkamp, M. (2015). Evolution of dynamin: Modular design of a membrane remodeling machine. BioEssays, 37(4), 348. https://doi.org/10.1002/bies.201400197
- Crewe, C., Joffin, N., Rutkowski, J. M., Kim, M., Zhang, F., Towler, D. A., Gordillo, R., & Scherer, P. E. (2018). An endothelial-to-adipocyte extracellular vesicle axis governed by metabolic state. Cell, 175(3), 695–708.e13. https://doi.org/10.1016/j.cell.2018.09.005
- Mathiesen, A., Hamilton, T., Carter, N., Brown, M., McPheat, W., & Dobrian, A. (2021). Endothelial extracellular vesicles: From keepers of health to messengers of disease. International Journal of Molecular Sciences, 22(9), 4640. https://doi.org/10.3390/ijms22094640
- Murakami, K., Tenge, V. R., Karandikar, U. C., Lin, S. C., Ramani, S., Ettayebi, K., Crawford, S. E., Zeng, X. L., Neill, F. H., Ayyar, B. V., Katayama, K., Graham, D. Y., Bieberich, E., Atmar, R. L., & Estes, M. K. (2020). Bile acids and ceramide overcome the entry restriction for GII.3 human norovirus replication. Proceedings of the National Academy of Sciences USA, 117(3), 1700–1710. https://doi.org/10.1073/pnas.1910138117
- Prichard, K. L., O'Brien, N. S., Murcia, S. R., Baker, J. R., & McCluskey, A. (2022). Role of clathrin and dynamin in clathrin-mediated endocytosis/synaptic vesicle recycling and implications in neurological diseases. Frontiers in Cellular Neuroscience, 15, 754110. https://doi.org/10.3389/fncel.2021.754110
- Shivanna, V., Kim, Y., & Chang, K. O. (2015). Ceramide formation mediated by acid sphingomyelinase facilitates endosomal escape of caliciviruses. Virology, 483, 218–228. https://doi.org/10.1016/j.virol.2015.04.022
- Peng, W., Chen, S., Ma, J., et al. (2025). Endosomal trafficking participates in lipid droplet catabolism to maintain lipid homeostasis. Nature Communications, 16, 1917. https://doi.org/10.1038/s41467-025-57038-8
- Xiao, J., Dong, L. W., Liu, S., et al. (2023). Bile acids-mediated intracellular cholesterol transport promotes intestinal cholesterol absorption and NPC1L1 recycling. Nature Communications, 14, 6469. https://doi.org/10.1038/s41467-023-42179-5
- Ybe, J. A. (2014). Novel clathrin activity: Developments in health and disease. Biomolecular Concepts, 5(2), 175–182. https://doi.org/10.1515/bmc-2013-0040
GLA v2.9+ — Canonical framework
Current authoritative mechanistic models defining PEM as a recovery-phase failure.
GLA v2.9+ — Modules
Focused modules expanding Tier 1 hinge logic and Tier 2 timing architecture.
Framework documents
Core architecture and definitions that anchor the GLA model.
Papers
Longer, paper-format documents (reader narrative + figures).
Modules (v2.1 → v2.6)
Modular “building blocks” used across the site. Organized by version and topic.
SMPDL3B phenotype frameworks
Phenotype-specific models (shedding vs deficient) and the mechanistic chain framework.
System modulators & control-state modifiers
Documents that shape interpretation of the core framework and control-state behavior.